

Integrated epigenomic profiling reveals endogenous retrovirus reactivation in renal cell carcinoma
- PMID: 30827930
- PMCID: PMC6441874
- DOI: 10.1016/j.ebiom.2019.01.063
Integrated epigenomic profiling reveals endogenous retrovirus reactivation in renal cell carcinoma
Abstract
Background: Transcriptional dysregulation drives cancer formation but the underlying mechanisms are still poorly understood. Renal cell carcinoma (RCC) is the most common malignant kidney tumor which canonically activates the hypoxia-inducible transcription factor (HIF) pathway. Despite intensive study, novel therapeutic strategies to target RCC have been difficult to develop. Since the RCC epigenome is relatively understudied, we sought to elucidate key mechanisms underpinning the tumor phenotype and its clinical behavior.
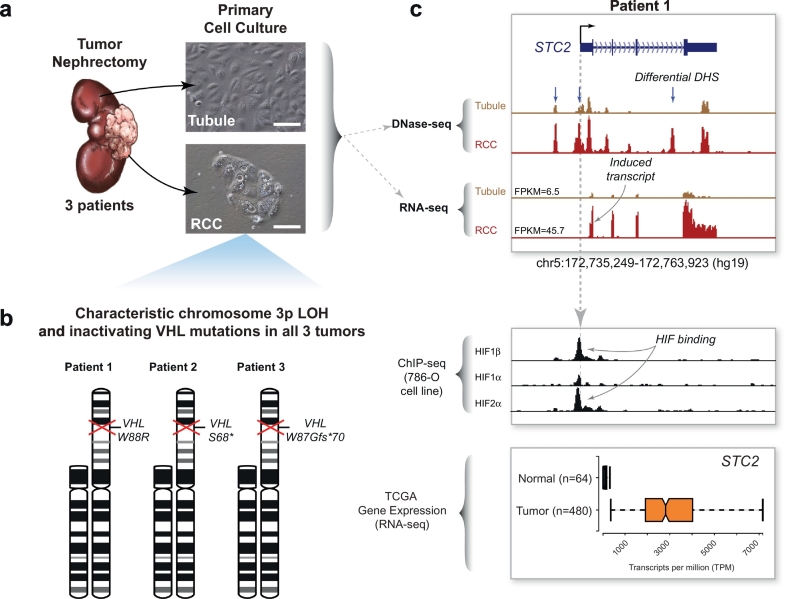
Methods: We performed genome-wide chromatin accessibility (DNase-seq) and transcriptome profiling (RNA-seq) on paired tumor/normal samples from 3 patients undergoing nephrectomy for removal of RCC. We incorporated publicly available data on HIF binding (ChIP-seq) in a RCC cell line. We performed integrated analyses of these high-resolution, genome-scale datasets together with larger transcriptomic data available through The Cancer Genome Atlas (TCGA).
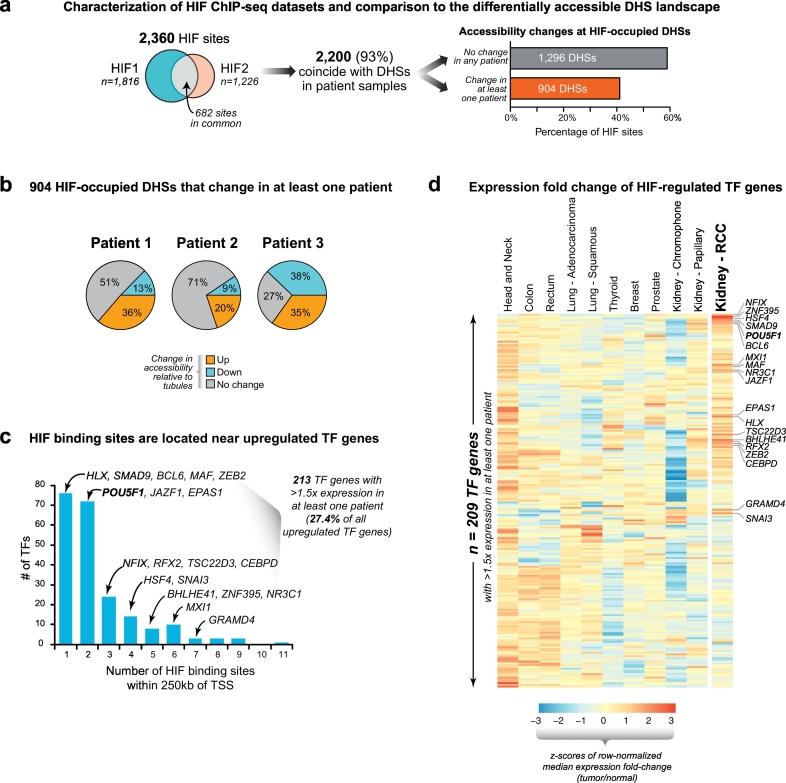
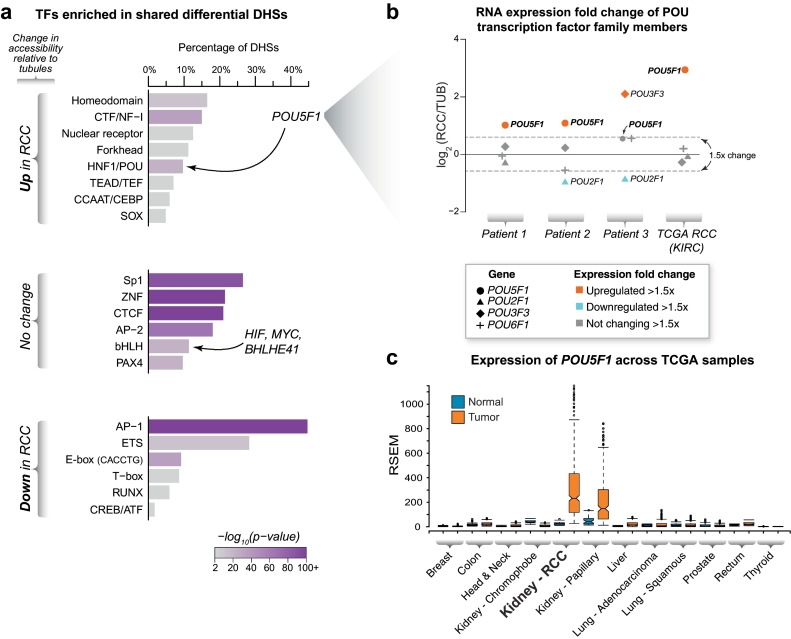
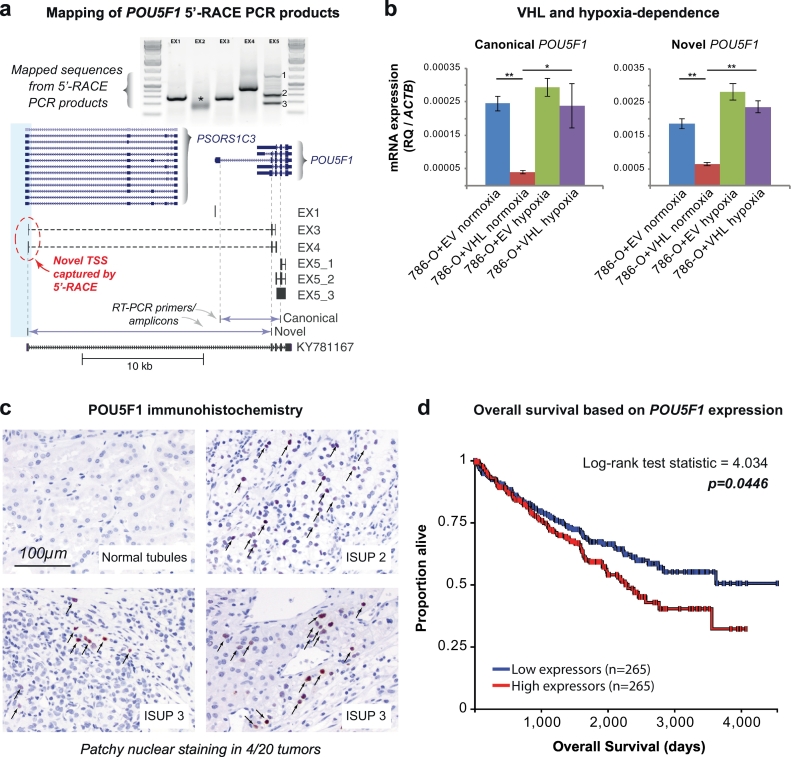
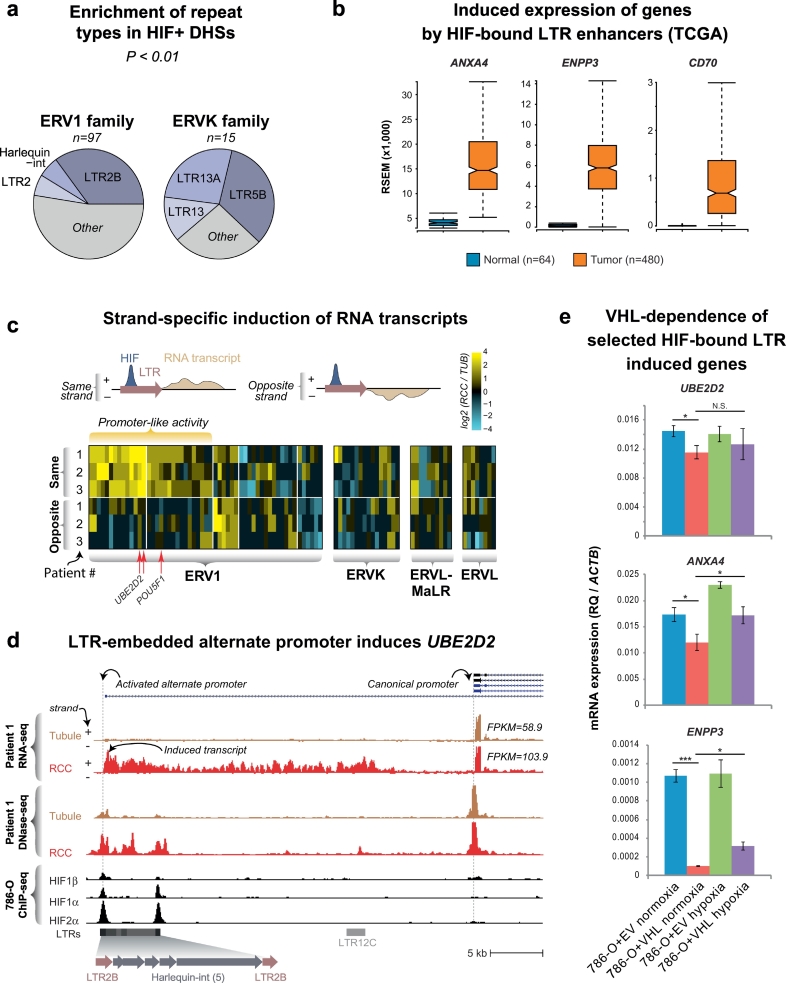
Findings: Though HIF transcription factors play a cardinal role in RCC oncogenesis, we found that numerous transcription factors with a RCC-selective expression pattern also demonstrated evidence of HIF binding near their gene body. Examination of chromatin accessibility profiles revealed that some of these transcription factors influenced the tumor's regulatory landscape, notably the stem cell transcription factor POU5F1 (OCT4). Elevated POU5F1 transcript levels were correlated with advanced tumor stage and poorer overall survival in RCC patients. Unexpectedly, we discovered a HIF-pathway-responsive promoter embedded within a endogenous retroviral long terminal repeat (LTR) element at the transcriptional start site of the PSOR1C3 long non-coding RNA gene upstream of POU5F1. RNA transcripts are induced from this promoter and read through PSOR1C3 into POU5F1 producing a novel POU5F1 transcript isoform. Rather than being unique to the POU5F1 locus, we found that HIF binds to several other transcriptionally active LTR elements genome-wide correlating with broad gene expression changes in RCC.
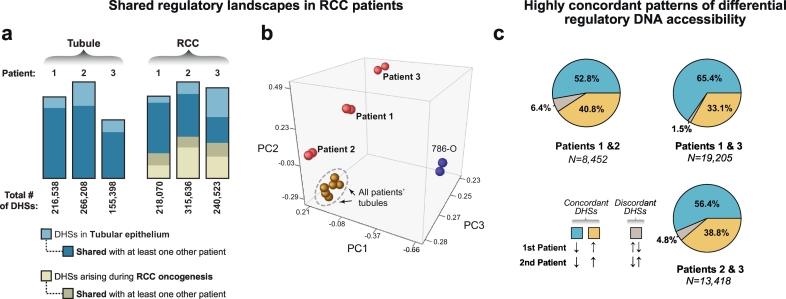
Interpretation: Integrated transcriptomic and epigenomic analysis of matched tumor and normal tissues from even a small number of primary patient samples revealed remarkably convergent shared regulatory landscapes. Several transcription factors appear to act downstream of HIF including the potent stem cell transcription factor POU5F1. Dysregulated expression of POU5F1 is part of a larger pattern of gene expression changes in RCC that may be induced by HIF-dependent reactivation of dormant promoters embedded within endogenous retroviral LTRs.
Keywords: Cancer epigenetics; Cancer stem cell; Kidney cancer; Regulatory genomics; Renal cell carcinoma; Transcription factors.
Copyright © 2019 The Authors. Published by Elsevier B.V. All rights reserved.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
