

Microbial abundance, activity and population genomic profiling with mOTUs2
- PMID: 30833550
- PMCID: PMC6399450
- DOI: 10.1038/s41467-019-08844-4
Microbial abundance, activity and population genomic profiling with mOTUs2
Abstract
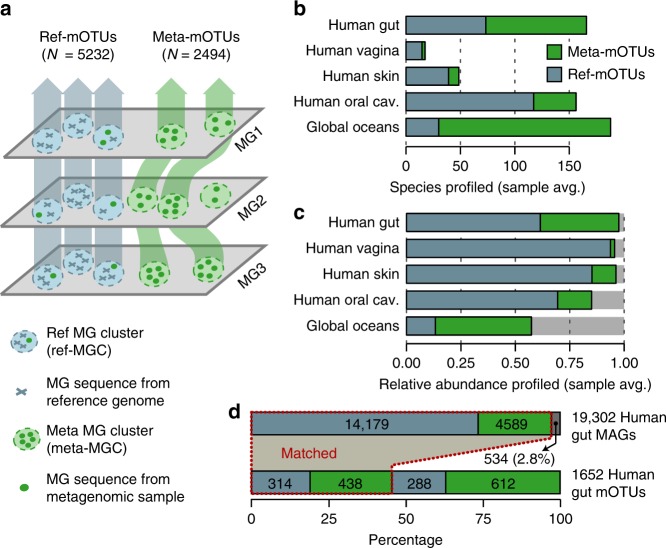
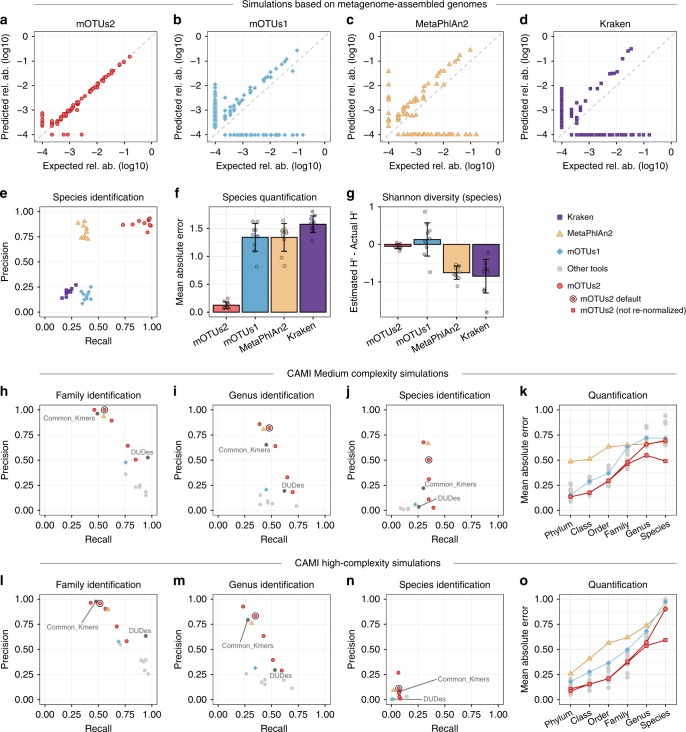
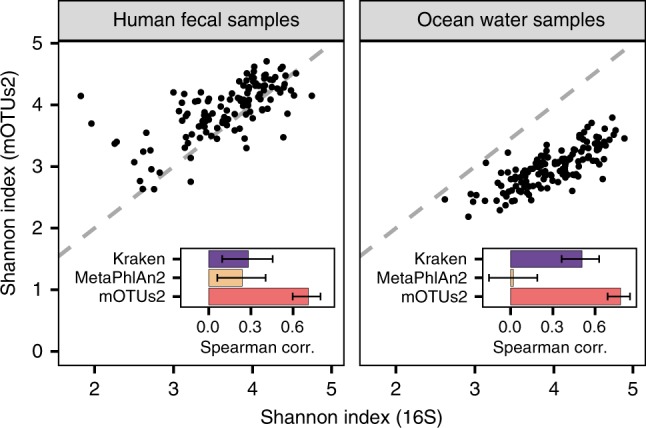
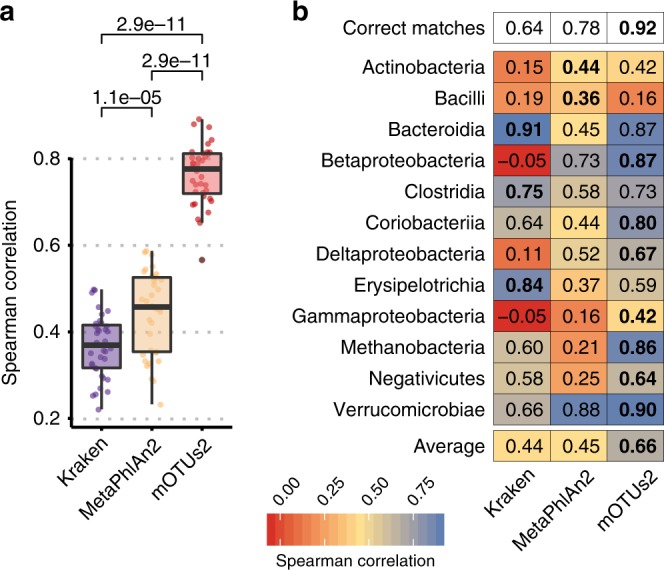
Metagenomic sequencing has greatly improved our ability to profile the composition of environmental and host-associated microbial communities. However, the dependency of most methods on reference genomes, which are currently unavailable for a substantial fraction of microbial species, introduces estimation biases. We present an updated and functionally extended tool based on universal (i.e., reference-independent), phylogenetic marker gene (MG)-based operational taxonomic units (mOTUs) enabling the profiling of >7700 microbial species. As more than 30% of them could not previously be quantified at this taxonomic resolution, relative abundance estimates based on mOTUs are more accurate compared to other methods. As a new feature, we show that mOTUs, which are based on essential housekeeping genes, are demonstrably well-suited for quantification of basal transcriptional activity of community members. Furthermore, single nucleotide variation profiles estimated using mOTUs reflect those from whole genomes, which allows for comparing microbial strain populations (e.g., across different human body sites).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Apprill A, McNally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015;75:129–137. doi: 10.3354/ame01753. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
