

Identification and genetic characterization of a novel Orthobunyavirus species by a straightforward high-throughput sequencing-based approach
- PMID: 30833612
- PMCID: PMC6399452
- DOI: 10.1038/s41598-019-40036-4
Identification and genetic characterization of a novel Orthobunyavirus species by a straightforward high-throughput sequencing-based approach
Abstract
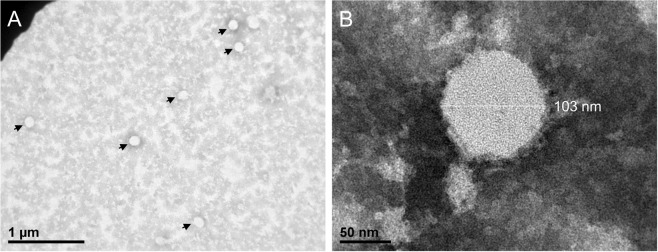
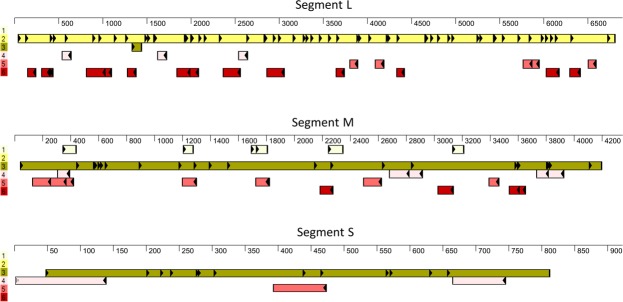
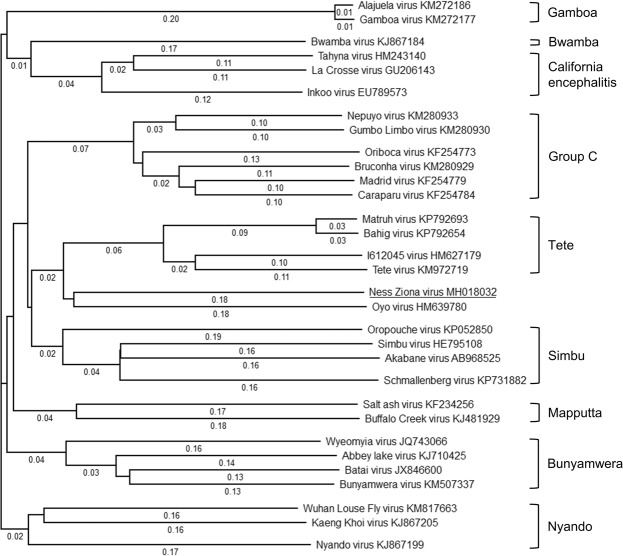
Identification and characterization of novel unknown viruses is of great importance. The introduction of high-throughput sequencing (HTS)-based methods has paved the way for genomics-based detection of pathogens without any prior assumptions about the characteristics of the organisms. However, the use of HTS for the characterization of viral pathogens from clinical samples remains limited. Here, we report the identification of a novel Orthobunyavirus species isolated from horse plasma. The identification was based on a straightforward HTS approach. Following enrichment in cell culture, RNA was extracted from the growth medium and rapid library preparation, HTS and primary bioinformatic analyses were performed in less than 12 hours. Taxonomical profiling of the sequencing reads did not reveal sequence similarities to any known virus. Subsequent application of de novo assembly tools to the sequencing reads produced contigs, of which three showed some similarity to the L, M, and S segments of viruses belonging to the Orthobunyavirus genus. Further refinement of these contigs resulted in high-quality, full-length genomic sequences of the three genomic segments (L, M and S) of a novel Orthobunyavirus. Characterization of the genomic sequence, including the prediction of open reading frames and the inspection of consensus genomic termini and phylogenetic analysis, further confirmed that the novel virus is indeed a new species, which we named Ness Ziona virus.
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
Molecular characterization of the African orthobunyavirus Ilesha virus.Infect Genet Evol. 2013 Dec;20:124-30. doi: 10.1016/j.meegid.2013.08.015. Epub 2013 Aug 26. Infect Genet Evol. 2013. PMID: 23988729
-
Random amplification and pyrosequencing for identification of novel viral genome sequences.J Biomol Tech. 2012 Apr;23(1):4-10. doi: 10.7171/jbt.12-2301-001. J Biomol Tech. 2012. PMID: 22468136 Free PMC article.
-
Molecular characterization of medically important viruses of the genus Orthobunyavirus.J Gen Virol. 2008 Oct;89(Pt 10):2580-2585. doi: 10.1099/vir.0.2008/002253-0. J Gen Virol. 2008. PMID: 18796727
-
Bioinformatics tools for analysing viral genomic data.Rev Sci Tech. 2016 Apr;35(1):271-85. doi: 10.20506/rst.35.1.2432. Rev Sci Tech. 2016. PMID: 27217183 Review.
-
Next generation sequencing technologies for insect virus discovery.Viruses. 2011 Oct;3(10):1849-69. doi: 10.3390/v3101849. Epub 2011 Oct 10. Viruses. 2011. PMID: 22069519 Free PMC article. Review.
Cited by
-
Metatranscriptomics to characterize respiratory virome, microbiome, and host response directly from clinical samples.Cell Rep Methods. 2021 Oct 25;1(6):100091. doi: 10.1016/j.crmeth.2021.100091. Cell Rep Methods. 2021. PMID: 34790908 Free PMC article.
-
Annual (2023) taxonomic update of RNA-directed RNA polymerase-encoding negative-sense RNA viruses (realm Riboviria: kingdom Orthornavirae: phylum Negarnaviricota).J Gen Virol. 2023 Aug;104(8):001864. doi: 10.1099/jgv.0.001864. J Gen Virol. 2023. PMID: 37622664 Free PMC article.
References
-
- Kapgate SS, Barbuddhe SB, Kumanan K. Acta Virol. 2015. Next generation sequencing technologies: tool to study avian virus diversity; pp. 3–13. - PubMed
-
- Storch, G. A. & Wang, D. In Fields Virology: Sixth Edition Vol. 1 (Wolters Kluwer Health Adis (ESP), 2013).
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
