

A constitutive knockout of murine carbamoyl phosphate synthetase 1 results in death with marked hyperglutaminemia and hyperammonemia
- PMID: 30835861
- PMCID: PMC6728231
- DOI: 10.1002/jimd.12048
A constitutive knockout of murine carbamoyl phosphate synthetase 1 results in death with marked hyperglutaminemia and hyperammonemia
Abstract
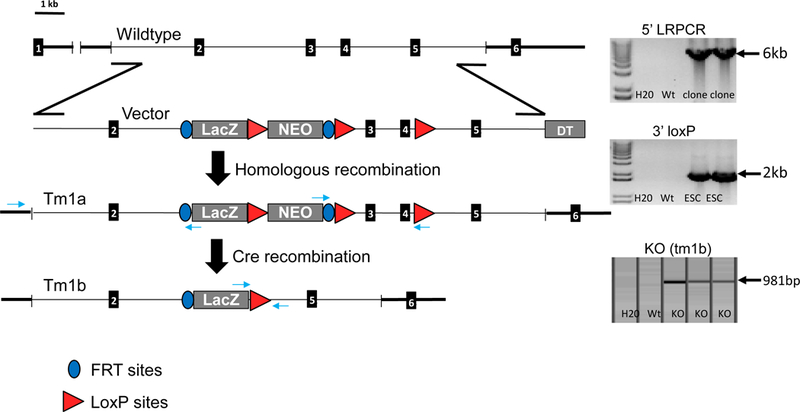
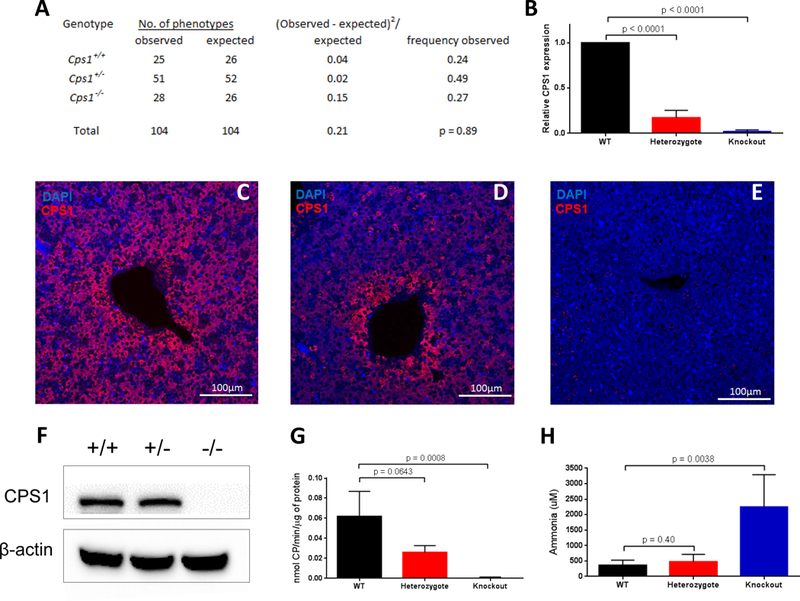
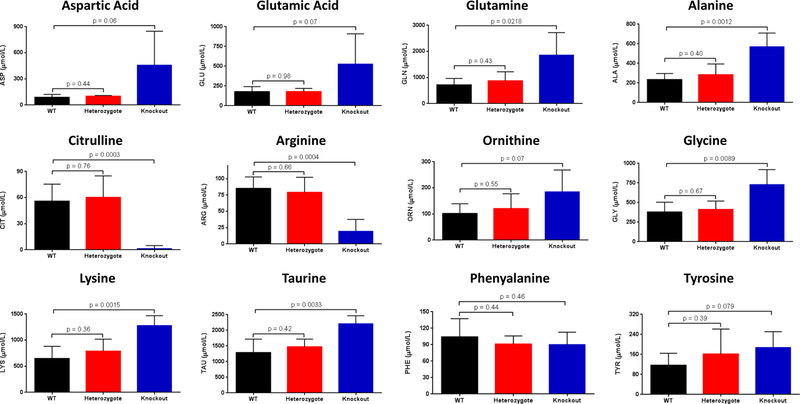
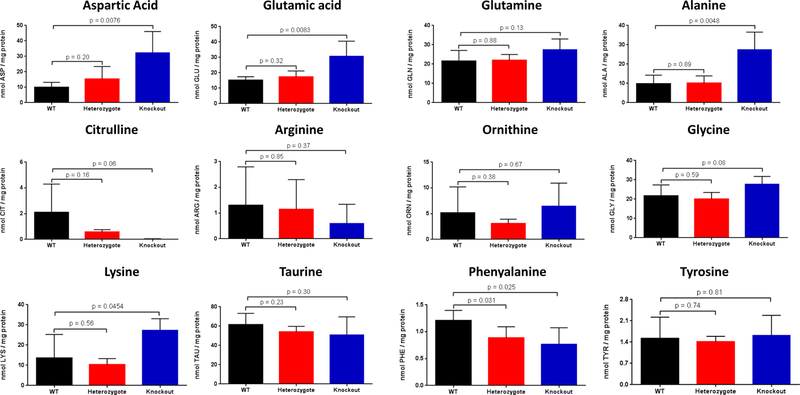
The enzyme carbamoyl phosphate synthetase 1 (CPS1; EC 6.3.4.16) forms carbamoyl phosphate from bicarbonate, ammonia, and adenosine triphosphate (ATP) and is activated allosterically by N-acetylglutamate. The neonatal presentation of bi-allelic mutations of CPS1 results in hyperammonemia with reduced citrulline and is reported as the most challenging nitrogen metabolism disorder to treat. As therapeutic interventions are limited, patients often develop neurological injury or die from hyperammonemia. Survivors remain vulnerable to nitrogen overload, being at risk for repetitive neurological injury. With transgenic technology, our lab developed a constitutive Cps1 mutant mouse and reports its characterization herein. Within 24 hours of birth, all Cps1 -/- mice developed hyperammonemia and expired. No CPS1 protein by Western blot or immunostaining was detected in livers nor was Cps1 mRNA present. CPS1 enzymatic activity was markedly decreased in knockout livers and reduced in Cps1+/- mice. Plasma analysis found markedly reduced citrulline and arginine and markedly increased glutamine and alanine, both intermolecular carriers of nitrogen, along with elevated ammonia, taurine, and lysine. Derangements in multiple other amino acids were also detected. While hepatic amino acids also demonstrated markedly reduced citrulline, arginine, while decreased, was not statistically significant; alanine and lysine were markedly increased while glutamine was trending towards significance. In conclusion we have determined that this constitutive neonatal mouse model of CPS1 deficiency replicates the neonatal human phenotype and demonstrates the key biochemical features of the disorder. These mice will be integral for addressing the challenges of developing new therapeutic approaches for this, at present, poorly treated disorder.
Keywords: carbamoyl phosphate synthetase (CPS1) deficiency; citrulline; glutamine; hyperammonemia; murine model.
© 2019 SSIEM.
Conflict of interest statement
Conflicts of Interest: Suhail Khoja, Matthew Nitzahn, Brian Truong, Jenna Lambert, Brandon Willis, Gabriella Allegri, Véronique Rüfenacht, Johannes Häberle declare that they have no conflict of interest. Gerald S. Lipshutz serves as a consultant to Audentes Therapeutics in a role unrelated to the studies conducted herein.
Conflict of Interest
The authors claim no conflict of interest.
Figures
References
-
- Ah Mew N, Lanpher B, Gropman AL, Chapman KA, Simpson KL, Summar ML (2003–2015) Urea Cycle Disorders Overview. In Editor ed. eds. Book Urea Cycle Disorders Overview Seattle, WA: University of Washington.
-
- Batshaw ML, Brusilow S, Waber L, et al. (1982) Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med 306: 1387–1392. - PubMed
-
- Batshaw ML, MacArthur RB, Tuchman M (2001) Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 138: S46–54; discussion S54–45. - PubMed
-
- Brosnan JT (1987) The 1986 Borden award lecture. The role of the kidney in amino acid metabolism and nutrition. Can J Physiol Pharmacol 65: 2355–2362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
