

A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression
- PMID: 30837243
- PMCID: PMC6497553
- DOI: 10.1158/2159-8290.CD-18-1212
A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression
Abstract
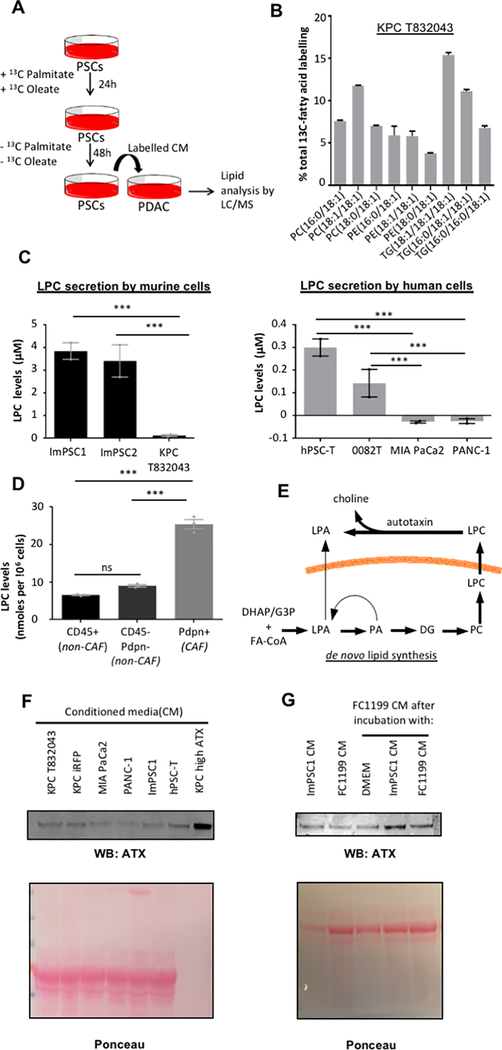
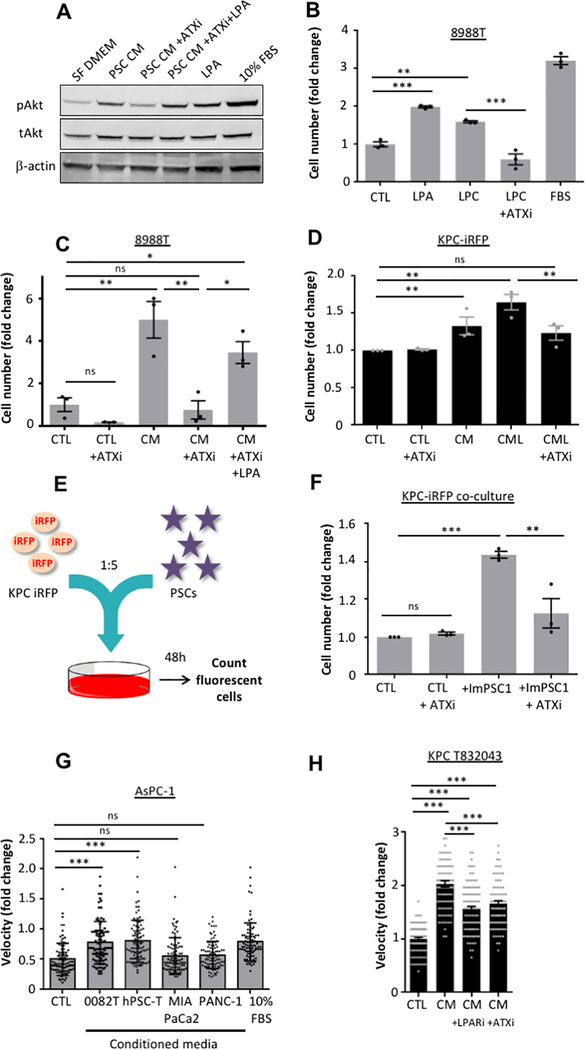
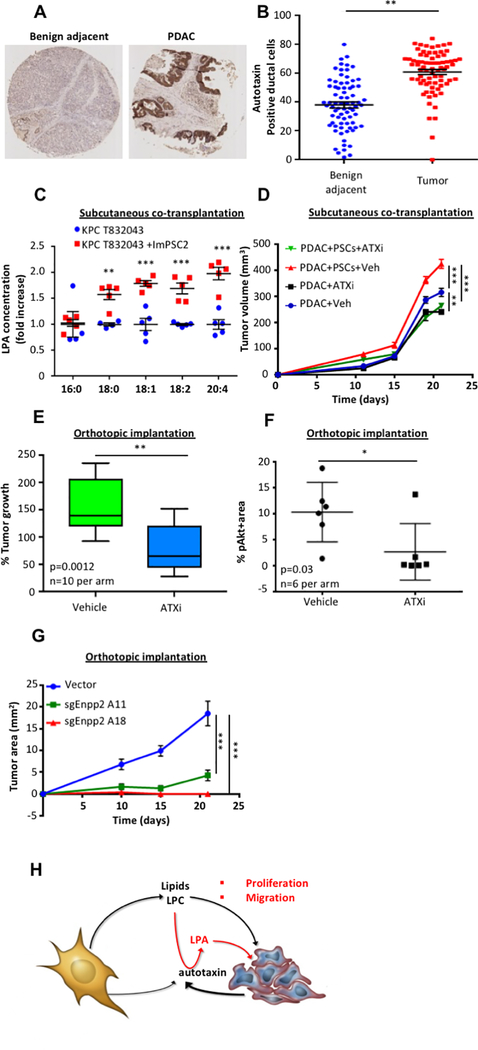
Pancreatic ductal adenocarcinoma (PDAC) develops a pronounced stromal response reflecting an aberrant wound-healing process. This stromal reaction features transdifferentiation of tissue-resident pancreatic stellate cells (PSC) into activated cancer-associated fibroblasts, a process induced by PDAC cells but of unclear significance for PDAC progression. Here, we show that PSCs undergo a dramatic lipid metabolic shift during differentiation in the context of pancreatic tumorigenesis, including remodeling of the intracellular lipidome and secretion of abundant lipids in the activated, fibroblastic state. Specifically, stroma-derived lysophosphatidylcholines support PDAC cell synthesis of phosphatidylcholines, key components of cell membranes, and also facilitate production of the potent wound-healing mediator lysophosphatidic acid (LPA) by the extracellular enzyme autotaxin, which is overexpressed in PDAC. The autotaxin-LPA axis promotes PDAC cell proliferation, migration, and AKT activation, and genetic or pharmacologic autotaxin inhibition suppresses PDAC growth in vivo. Our work demonstrates how PDAC cells exploit the local production of wound-healing mediators to stimulate their own growth and migration. SIGNIFICANCE: Our work highlights an unanticipated role for PSCs in producing the oncogenic LPA signaling lipid and demonstrates how PDAC tumor cells co-opt the release of wound-healing mediators by neighboring PSCs to promote their own proliferation and migration.See related commentary by Biffi and Tuveson, p. 578.This article is highlighted in the In This Issue feature, p. 565.
©2019 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest disclosure statement: JJK is an employee and shareholder of Rheos Medicines Inc.
Figures

Comment in
-
A FATal Combination: Fibroblast-Derived Lipids and Cancer-Derived Autotaxin Promote Pancreatic Cancer Growth.Cancer Discov. 2019 May;9(5):578-580. doi: 10.1158/2159-8290.CD-19-0273. Cancer Discov. 2019. PMID: 31043410
References
-
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155. PubMed PMID: . - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- C50242/A17728/CRUK_/Cancer Research UK/United Kingdom
- 29996/CRUK_/Cancer Research UK/United Kingdom
- C596/A18076/CRUK_/Cancer Research UK/United Kingdom
- R01 CA186241/CA/NCI NIH HHS/United States
- R01 CA196228/CA/NCI NIH HHS/United States
- 18277/CRUK_/Cancer Research UK/United Kingdom
- R00 CA188259/CA/NCI NIH HHS/United States
- K99 CA188259/CA/NCI NIH HHS/United States
- 28291/CRUK_/Cancer Research UK/United Kingdom
- R01 CA229580/CA/NCI NIH HHS/United States
- C596/A17196/CRUK_/Cancer Research UK/United Kingdom
- MR/P01058X/1/MRC_/Medical Research Council/United Kingdom
- P30 CA014195/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
