

The use of technical replication for detection of low-level somatic mutations in next-generation sequencing
- PMID: 30837471
- PMCID: PMC6400950
- DOI: 10.1038/s41467-019-09026-y
The use of technical replication for detection of low-level somatic mutations in next-generation sequencing
Abstract
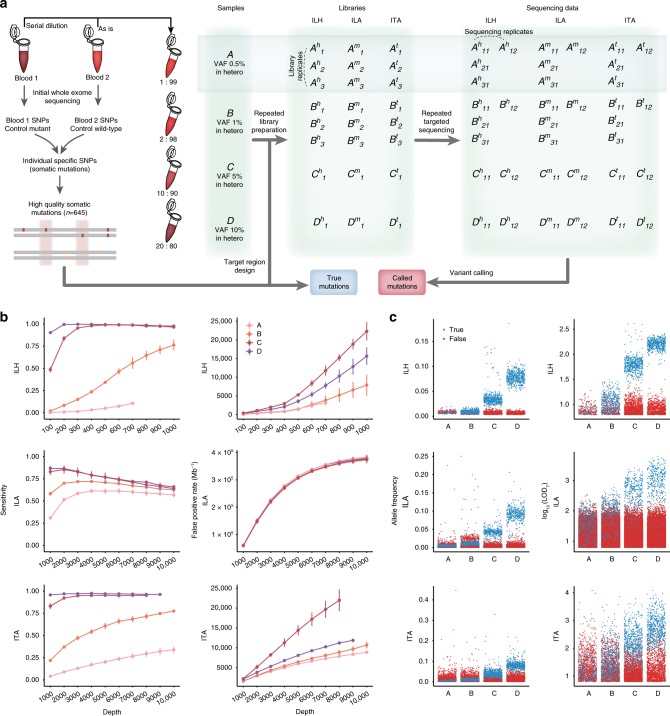
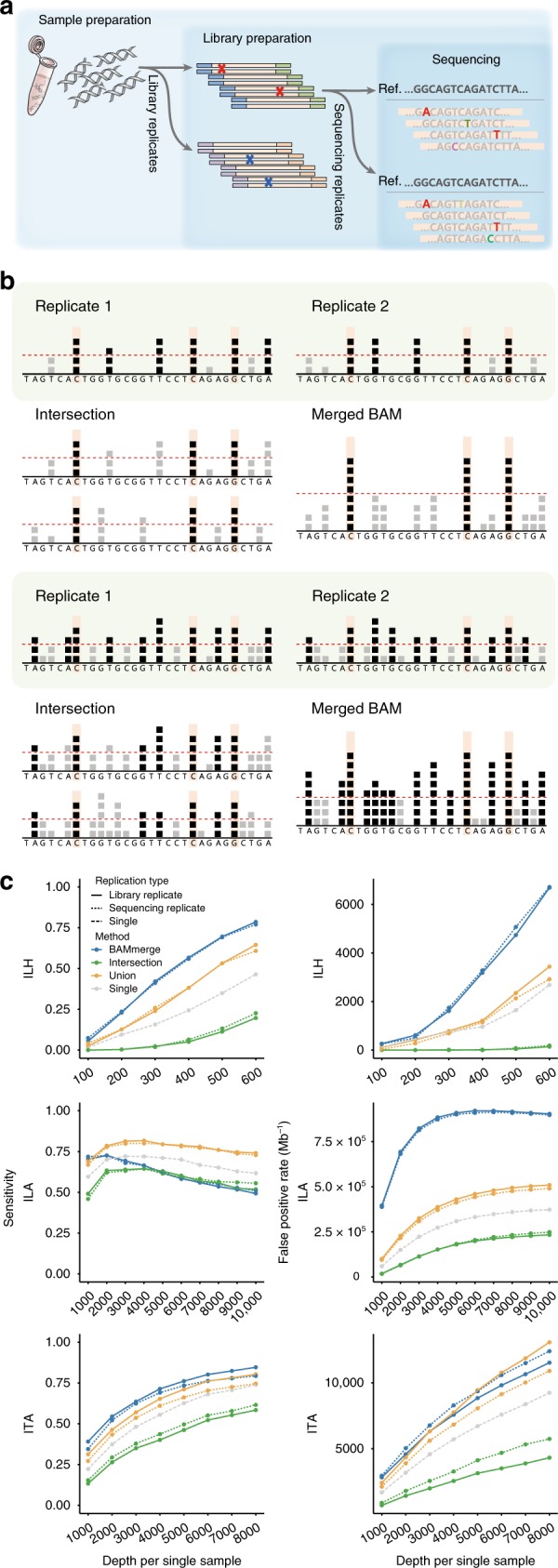
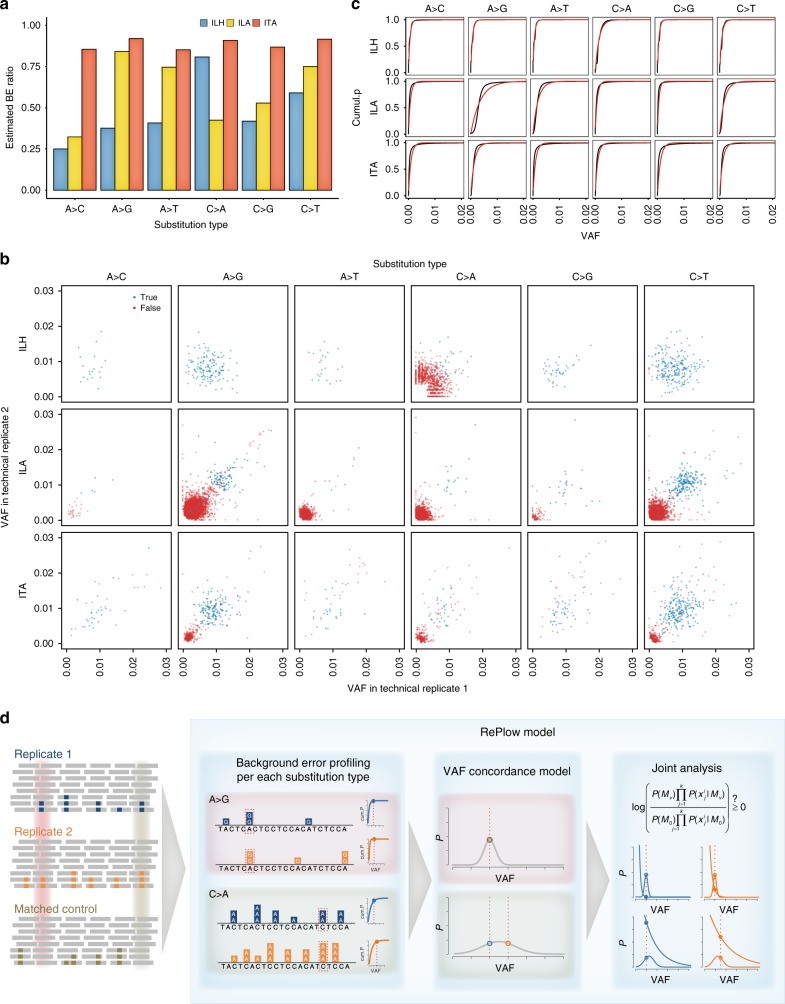
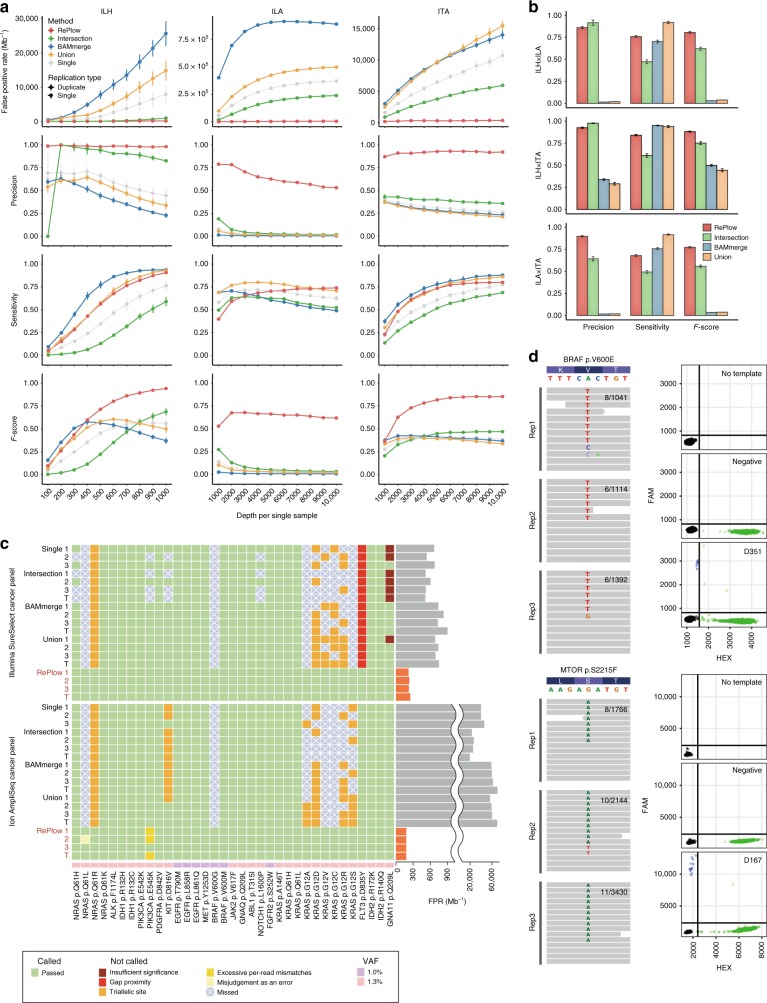
Accurate genome-wide detection of somatic mutations with low variant allele frequency (VAF, <1%) has proven difficult, for which generalized, scalable methods are lacking. Herein, we describe a new computational method, called RePlow, that we developed to detect low-VAF somatic mutations based on simple, library-level replicates for next-generation sequencing on any platform. Through joint analysis of replicates, RePlow is able to remove prevailing background errors in next-generation sequencing analysis, facilitating remarkable improvement in the detection accuracy for low-VAF somatic mutations (up to ~99% reduction in false positives). The method is validated in independent cancer panel and brain tissue sequencing data. Our study suggests a new paradigm with which to exploit an overwhelming abundance of sequencing data for accurate variant detection.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
