

Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis
- PMID: 30837838
- PMCID: PMC6382748
- DOI: 10.3389/fnmol.2019.00025
Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis
Abstract
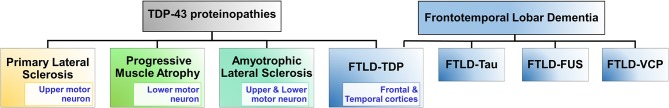
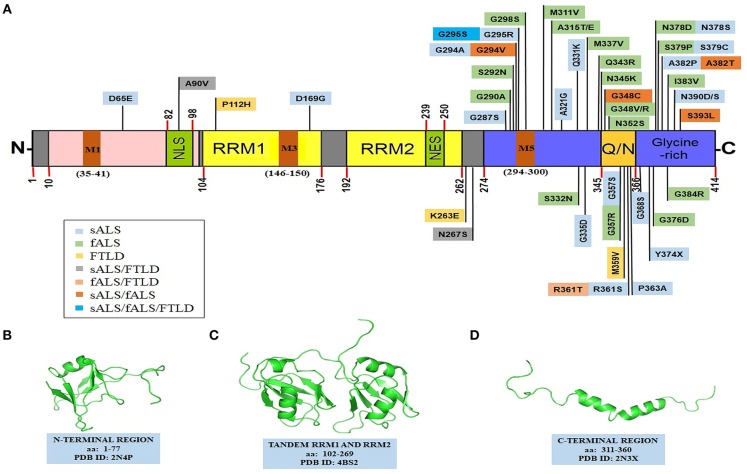
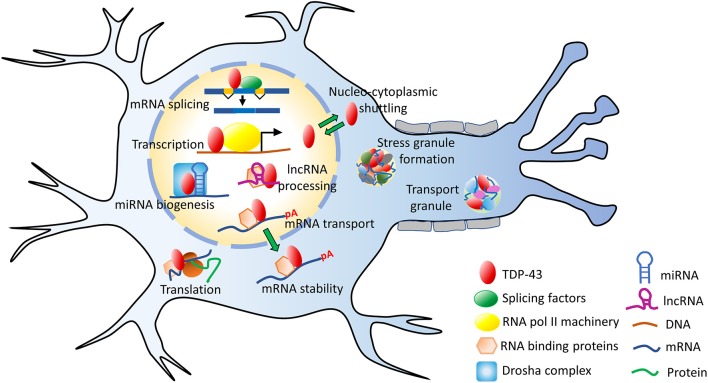
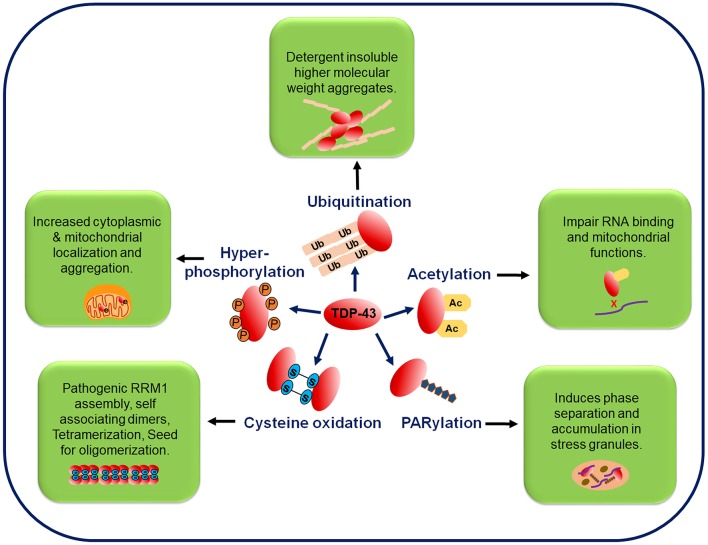
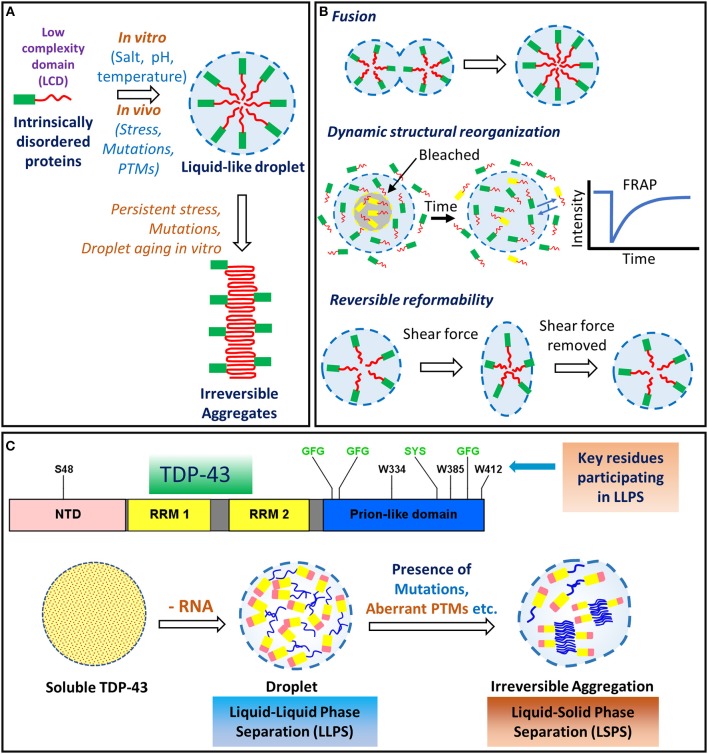
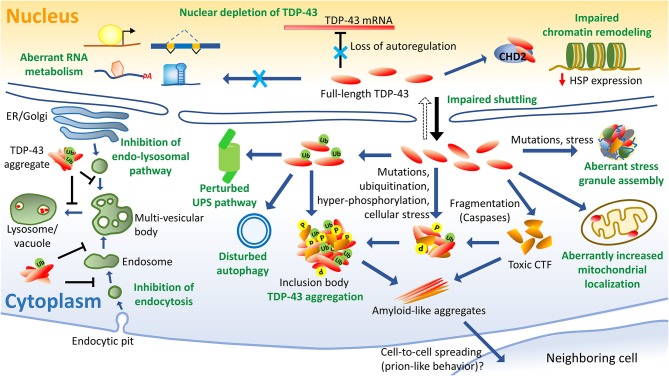
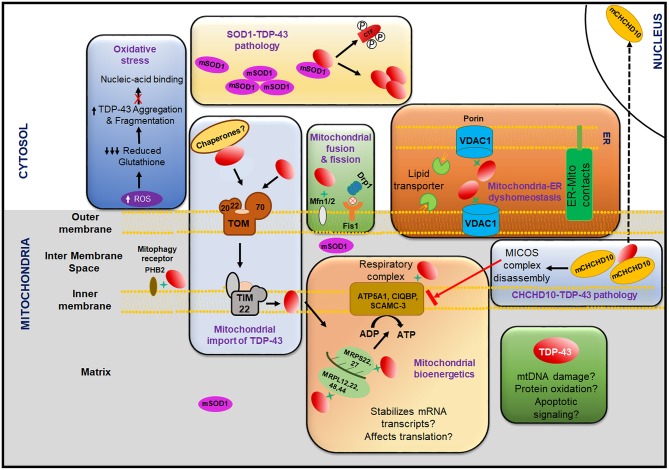
TAR DNA binding protein 43 (TDP-43) is a versatile RNA/DNA binding protein involved in RNA-related metabolism. Hyper-phosphorylated and ubiquitinated TDP-43 deposits act as inclusion bodies in the brain and spinal cord of patients with the motor neuron diseases: amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). While the majority of ALS cases (90-95%) are sporadic (sALS), among familial ALS cases 5-10% involve the inheritance of mutations in the TARDBP gene and the remaining (90-95%) are due to mutations in other genes such as: C9ORF72, SOD1, FUS, and NEK1 etc. Strikingly however, the majority of sporadic ALS patients (up to 97%) also contain the TDP-43 protein deposited in the neuronal inclusions, which suggests of its pivotal role in the ALS pathology. Thus, unraveling the molecular mechanisms of the TDP-43 pathology seems central to the ALS therapeutics, hence, we comprehensively review the current understanding of the TDP-43's pathology in ALS. We discuss the roles of TDP-43's mutations, its cytoplasmic mis-localization and aberrant post-translational modifications in ALS. Also, we evaluate TDP-43's amyloid-like in vitro aggregation, its physiological vs. pathological oligomerization in vivo, liquid-liquid phase separation (LLPS), and potential prion-like propagation propensity of the TDP-43 inclusions. Finally, we describe the various evolving TDP-43-induced toxicity mechanisms, such as the impairment of endocytosis and mitotoxicity etc. and also discuss the emerging strategies toward TDP-43 disaggregation and ALS therapeutics.
Keywords: ALS therapeutics; TDP-43; amyotrophic lateral sclerosis (ALS); endocytosis; frontotemporal lobar degeneration (FTLD); liquid-liquid phase separation (LLPS); mitotoxicity; prion.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous