

Oral Mucosal Epithelial Cells
- PMID: 30837987
- PMCID: PMC6383680
- DOI: 10.3389/fimmu.2019.00208
Oral Mucosal Epithelial Cells
Abstract
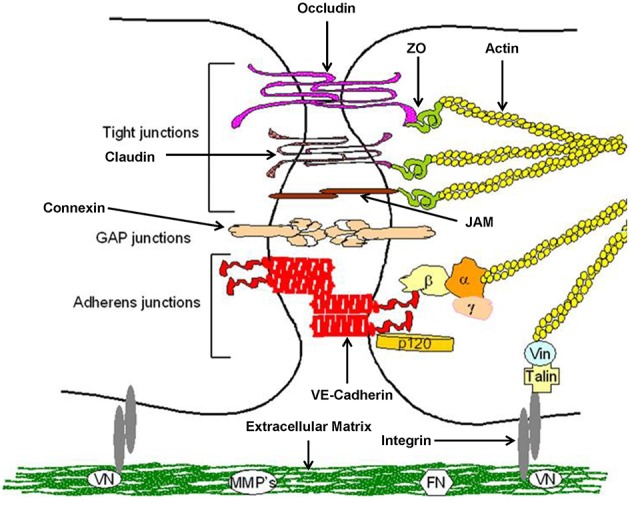
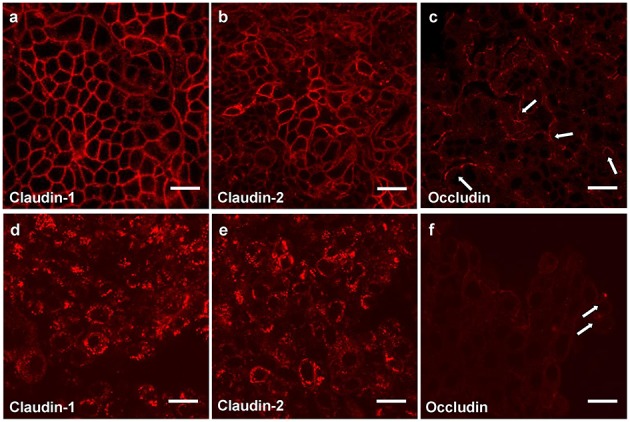
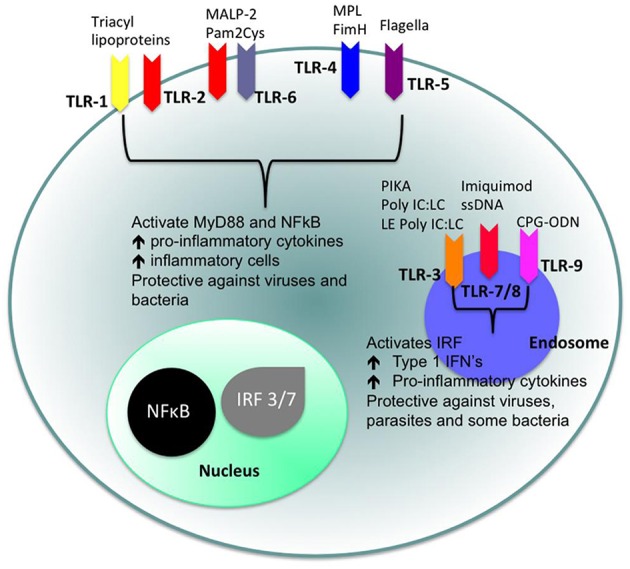
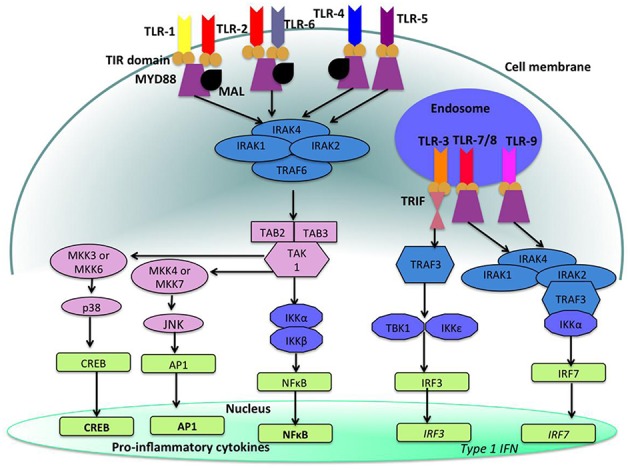
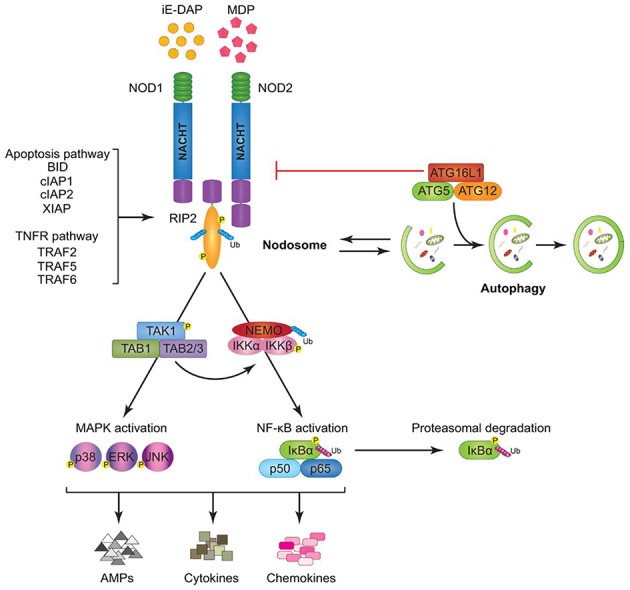
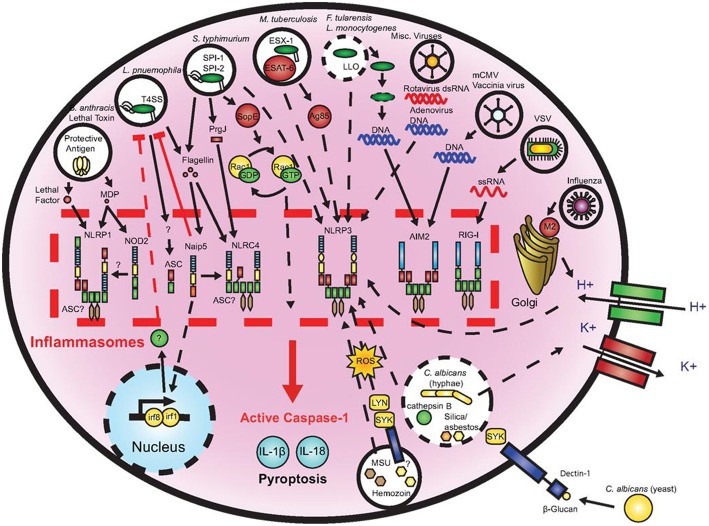
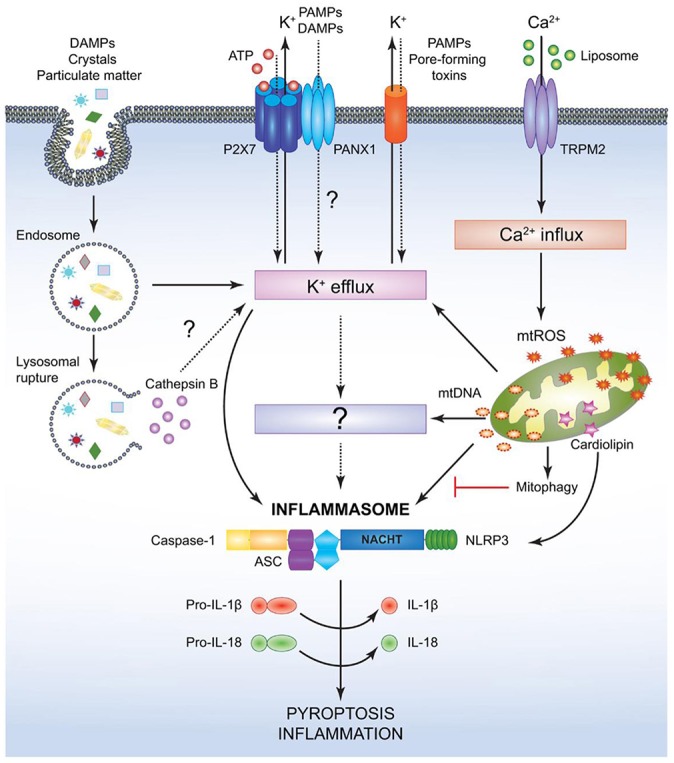
Cellular Phenotype and Apoptosis: The function of epithelial tissues is the protection of the organism from chemical, microbial, and physical challenges which is indispensable for viability. To fulfill this task, oral epithelial cells follow a strongly regulated scheme of differentiation that results in the formation of structural proteins that manage the integrity of epithelial tissues and operate as a barrier. Oral epithelial cells are connected by various transmembrane proteins with specialized structures and functions. Keratin filaments adhere to the plasma membrane by desmosomes building a three-dimensional matrix. Cell-Cell Contacts and Bacterial Influence: It is known that pathogenic oral bacteria are able to affect the expression and configuration of cell-cell junctions. Human keratinocytes up-regulate immune-modulatory receptors upon stimulation with bacterial components. Periodontal pathogens including P. gingivalis are able to inhibit oral epithelial innate immune responses through various mechanisms and to escape from host immune reaction, which supports the persistence of periodontitis and furthermore is able to affect the epithelial barrier function by altering expression and distribution of cell-cell interactions including tight junctions (TJs) and adherens junctions (AJs). In the pathogenesis of periodontitis a highly organized biofilm community shifts from symbiosis to dysbiosis which results in destructive local inflammatory reactions. Cellular Receptors: Cell-surface located toll like receptors (TLRs) and cytoplasmatic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) belong to the pattern recognition receptors (PRRs). PRRs recognize microbial parts that represent pathogen-associated molecular patterns (PAMPs). A multimeric complex of proteins known as inflammasome, which is a subset of NLRs, assembles after activation and proceeds to pro-inflammatory cytokine release. Cytokine Production and Release: Cytokines and bacterial products may lead to host cell mediated tissue destruction. Keratinocytes are able to produce diverse pro-inflammatory cytokines and chemokines, including interleukin (IL)-1, IL-6, IL-8 and tumor necrosis factor (TNF)-α. Infection by pathogenic bacteria such as Porphyromonas gingivalis (P. gingivalis) and Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans) can induce a differentiated production of these cytokines. Immuno-modulation, Bacterial Infection, and Cancer Cells: There is a known association between bacterial infection and cancer. Bacterial components are able to up-regulate immune-modulatory receptors on cancer cells. Interactions of bacteria with tumor cells could support malignant transformation an environment with deficient immune regulation. The aim of this review is to present a set of molecular mechanisms of oral epithelial cells and their reactions to a number of toxic influences.
Keywords: cancer; cytokines; differentiation; immuno-modulation; infection; oral epithelial cells; receptors.
Figures

References
-
- Gartner LP. Oral anatomy and tissue types. Semin Dermatol. (1994) 13:68–73. - PubMed
-
- Garant PR. Oral Cells and Tissues. Illinois, IL: Quintessence Publishing Co., Inc; (2003).
-
- Schroeder HE, Listgarten MA. The gingival tissues: the architecture of periodontal protection. Periodontol (1997)13:91–120. - PubMed
-
- Squier CA, Kremer MJ. Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr. (2001) 29:7–15. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources