

Liver Failure in a Chinese Cystic Fibrosis Child With Homozygous R553X Mutation
- PMID: 30842938
- PMCID: PMC6391319
- DOI: 10.3389/fped.2019.00036
Liver Failure in a Chinese Cystic Fibrosis Child With Homozygous R553X Mutation
Abstract
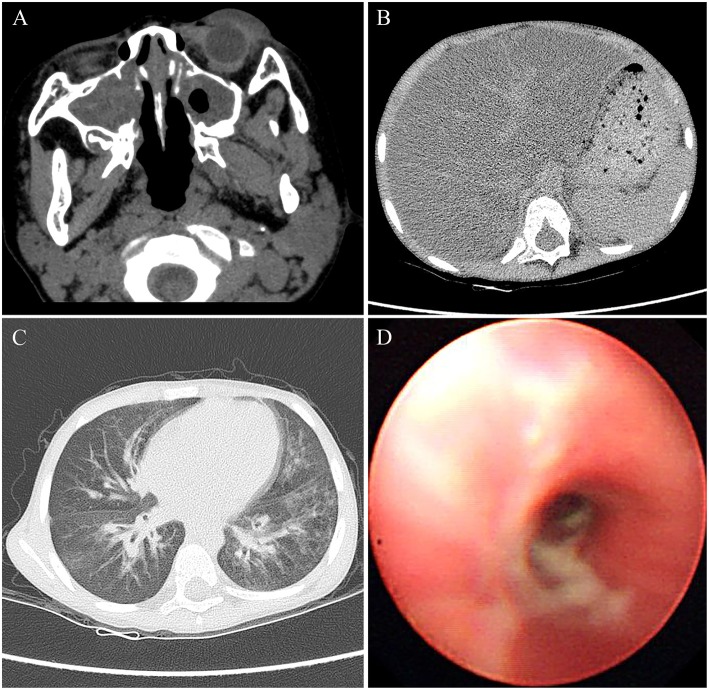
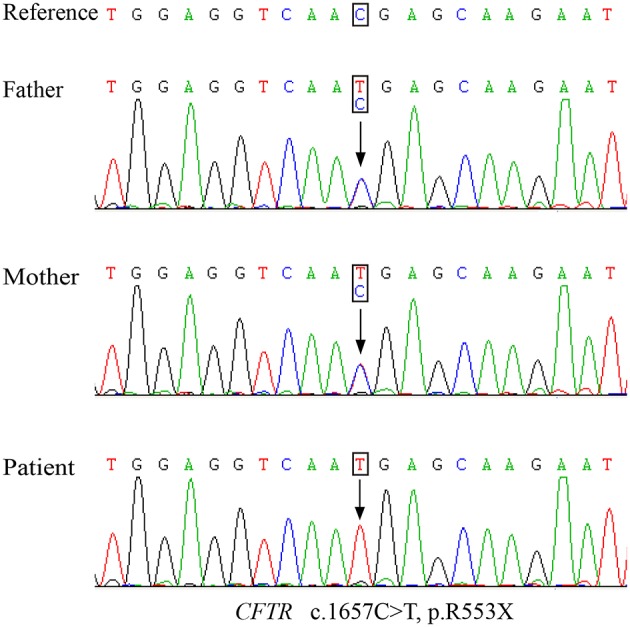
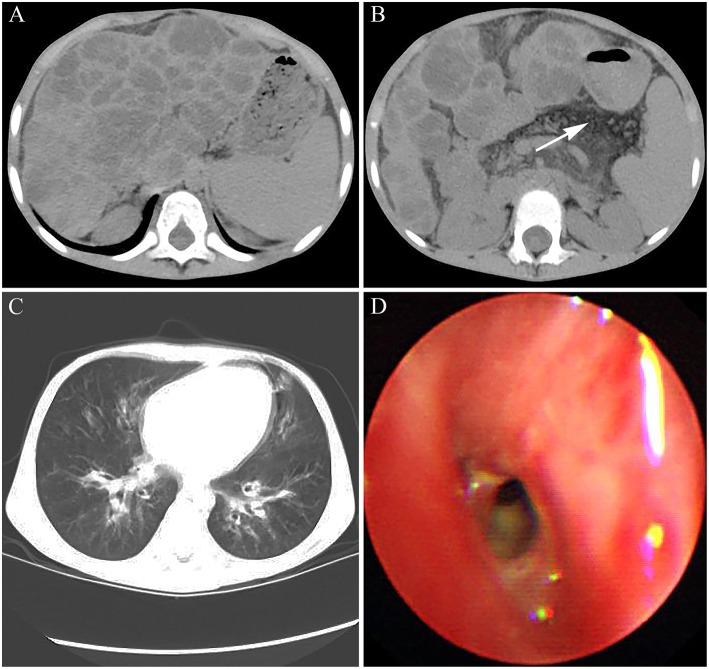
Cystic fibrosis (CF) is a relatively rare disease in Asians with various clinical characteristics, including CF-associated liver disease (CFLD), which is a common early non-pulmonary complication. This case report describes a Chinese CF patient harboring a homozygous nonsense mutation (c.1657C>T, p.R553X) who was failure to thrive and had intermittently diarrhea during the first year after birth. Liver function test of the patient showed the mildly and intermittently elevated alanine aminotransferase (ALT) levels ranging from 70 to 92 U/L and aspartate aminotransferase (AST) levels ranging from 80 to 90 U/L, which began at 8 months of age and lasted for 4 years without CF diagnosis. In addition, abdominal computed tomography (CT) revealed diffuse fatty infiltration of the liver at 4 years old and gradually developed hepatic cirrhosis. Subsequently, cirrhosis rapidly progressed with obvious splenomegaly and pancreatic insufficiency and the patient died of liver failure with coagulopathy by the age of 6 years old. Pediatricians should remain vigilant to avoid failure to diagnose CF, the occurrence of which may be underestimated, and pay greater attention to the patients with atypical clinical manifestations in Asian countries.
Keywords: CF transmembrane conductance regulator; CF-associated liver disease; cystic fibrosis; homozygous CFTR mutation; liver failure.
Figures
Similar articles
-
Severe deficiency of cystic fibrosis transmembrane conductance regulator messenger RNA carrying nonsense mutations R553X and W1316X in respiratory epithelial cells of patients with cystic fibrosis.J Clin Invest. 1991 Dec;88(6):1880-5. doi: 10.1172/JCI115510. J Clin Invest. 1991. PMID: 1721624 Free PMC article.
-
Cystic fibrosis with homozygous R553X mutation in a Taiwanese child.J Hum Genet. 2005;50(12):674-8. doi: 10.1007/s10038-005-0309-x. Epub 2005 Nov 10. J Hum Genet. 2005. PMID: 16283068
-
c.753_754delAG, a novel CFTR mutation found in a Chinese patient with cystic fibrosis: A case report and review of the literature.World J Clin Cases. 2019 Aug 6;7(15):2110-2119. doi: 10.12998/wjcc.v7.i15.2110. World J Clin Cases. 2019. PMID: 31423445 Free PMC article.
-
Cystic Fibrosis Liver Disease: Know More.Oman Med J. 2019 Nov;34(6):482-489. doi: 10.5001/omj.2019.90. Oman Med J. 2019. PMID: 31745411 Free PMC article. Review.
-
[A case report of cystic fibrosis and review of 16 cases of cystic fibrosis in Chinese patients].Zhonghua Jie He He Hu Xi Za Zhi. 2003 Sep;26(9):559-62. Zhonghua Jie He He Hu Xi Za Zhi. 2003. PMID: 14521762 Review. Chinese.
Cited by
-
Pediatric lung transplantation in the largest lung transplantation center of China: embarking on a long road.Sci Rep. 2020 Jul 27;10(1):12471. doi: 10.1038/s41598-020-69340-0. Sci Rep. 2020. PMID: 32719472 Free PMC article.
-
Characterization of clinical and genetic spectrum of Chinese patients with cystic fibrosis.Orphanet J Rare Dis. 2020 Jun 15;15(1):150. doi: 10.1186/s13023-020-01393-w. Orphanet J Rare Dis. 2020. PMID: 32539862 Free PMC article.
-
Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease.Cells. 2021 Aug 12;10(8):2072. doi: 10.3390/cells10082072. Cells. 2021. PMID: 34440841 Free PMC article. Review.
-
[Detection of pathogenic gene mutations in thirteen cases of congenital bilateral absence of vas deferens infertility patients].Beijing Da Xue Xue Bao Yi Xue Ban. 2024 Oct 18;56(5):763-774. doi: 10.19723/j.issn.1671-167X.2024.05.003. Beijing Da Xue Xue Bao Yi Xue Ban. 2024. PMID: 39397452 Free PMC article. Chinese.
-
Genetic spectrum of Chinese children with cystic fibrosis: comprehensive data analysis from the main referral centre in China.J Med Genet. 2022 Jul 20;60(3):310-5. doi: 10.1136/jmg-2022-108501. Online ahead of print. J Med Genet. 2022. PMID: 35858753 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources