

Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms
- PMID: 30843314
- PMCID: PMC6483869
- DOI: 10.1111/liv.14091
Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms
Abstract
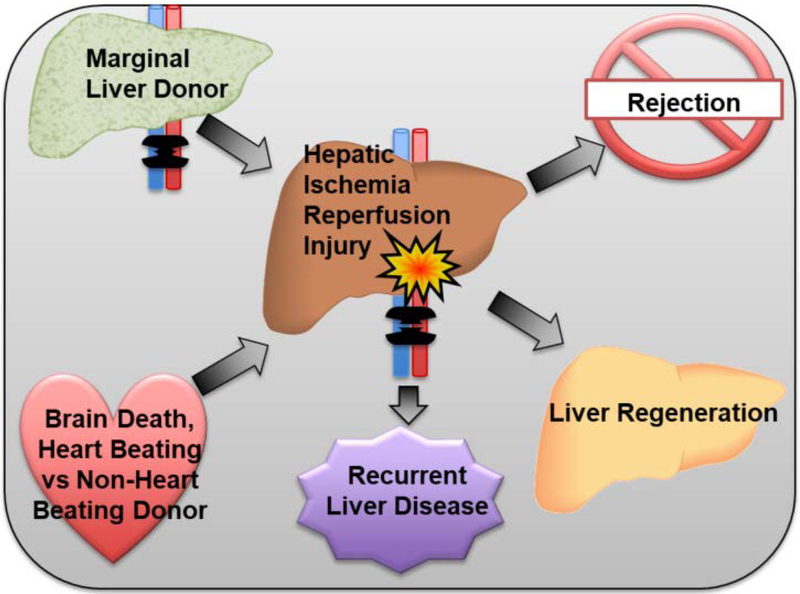
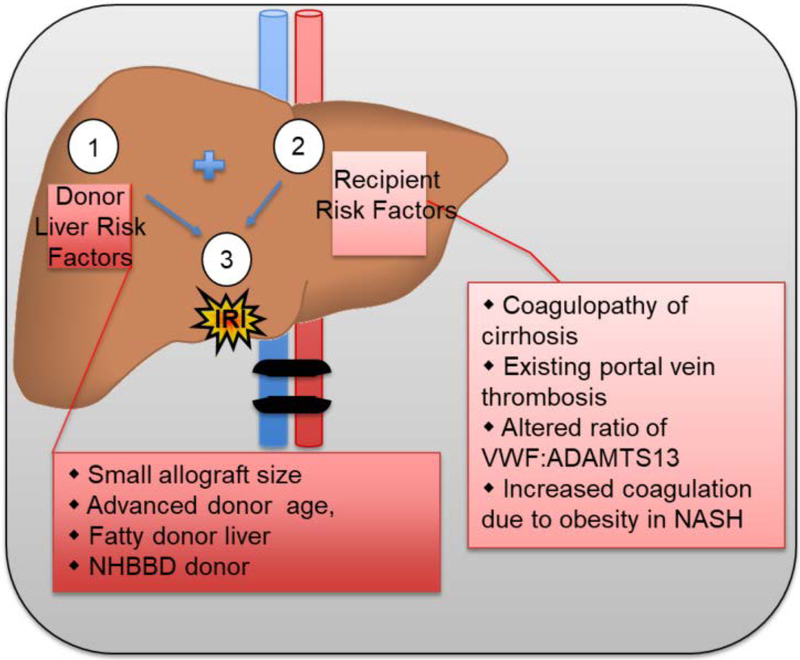
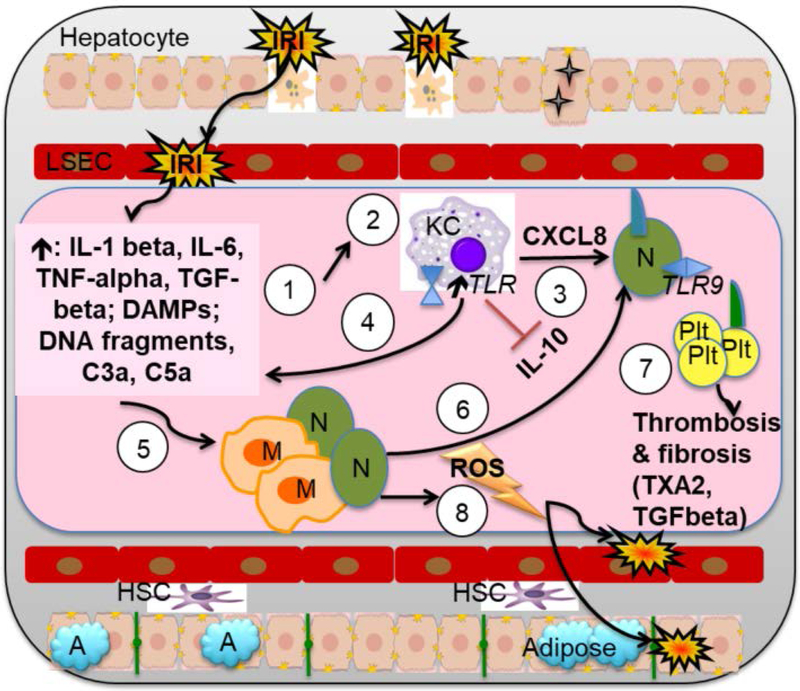
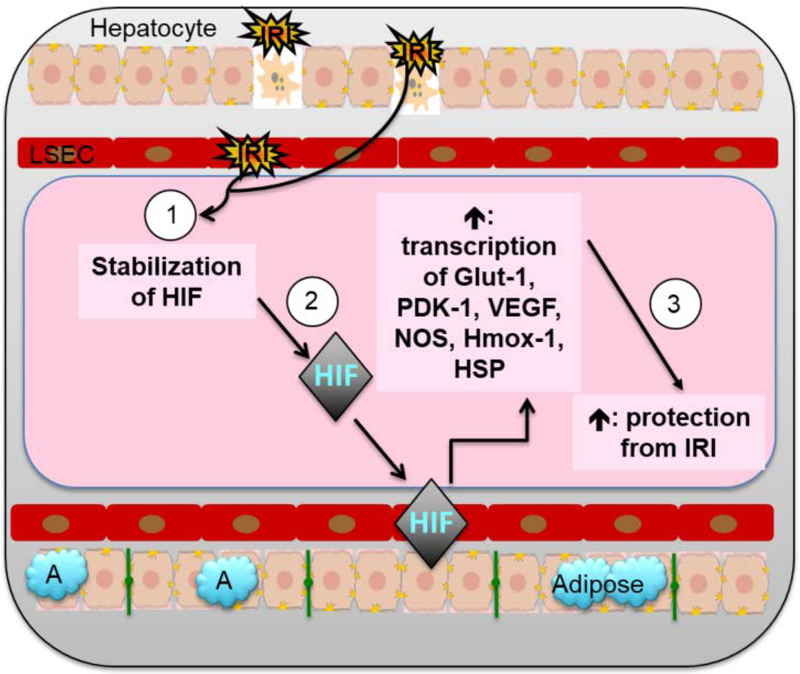
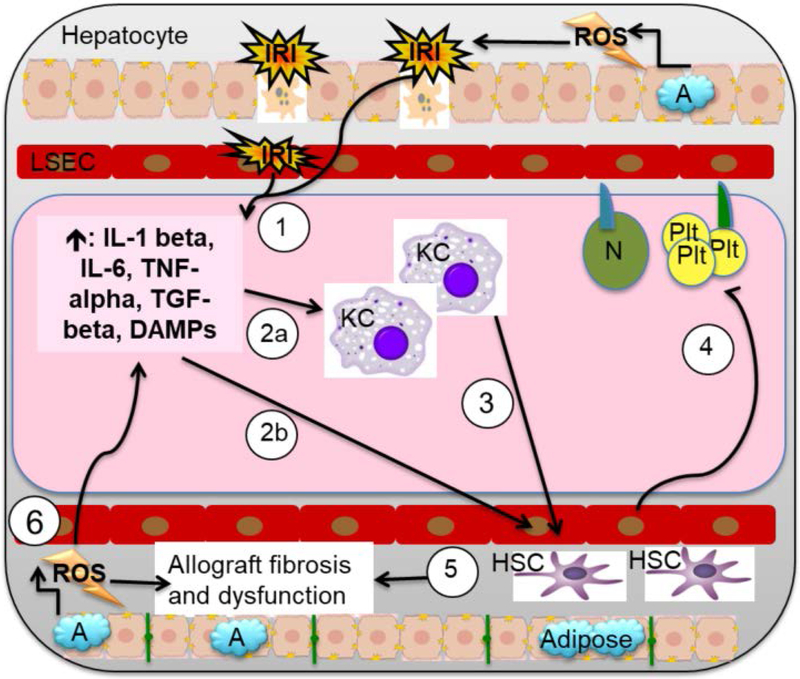
Liver disease causing end organ failure is a growing cause of mortality. In most cases, the only therapy is liver transplantation. However, liver transplantation is a complex undertaking and its success is dependent on a number of factors. In particular, liver transplantation is subject to the risks of ischaemia-reperfusion injury (IRI). Liver IRI has significant effects on the function of a liver after transplantation. The cellular and molecular mechanisms governing IRI in liver transplantation are numerous. They involve multiple cells types such as liver sinusoidal endothelial cells, hepatocytes, Kupffer cells, neutrophils and platelets acting via an interconnected network of molecular pathways such as activation of toll-like receptor signalling, alterations in micro-RNA expression, production of ROS, regulation of autophagy and activation of hypoxia-inducible factors. Interestingly, the cellular and molecular events in liver IRI can be correlated with clinical risk factors for IRI in liver transplantation such as donor organ steatosis, ischaemic times, donor age, and donor and recipient coagulopathy. Thus, understanding the relationship of the clinical risk factors for liver IRI to the cellular and molecular mechanisms that govern it is critical to higher levels of success after liver transplantation. This in turn will help in the discovery of therapeutics for IRI in liver transplantation - a process that will lead to improved outcomes for patients suffering from end-stage liver disease.
Keywords: hypoxia-inducible factors; ischaemia reperfusion; liver transplantation; therapeutics.
© 2019 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
COI: Authors have no conflict of interests.
Figures
References
-
- Marcellin P, Kutala BK. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int 2018; 38 Suppl 1: 2–6. - PubMed
-
- JENNINGS RB, SOMMERS HM, SMYTH GA, FLACK HA, LINN H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol 1960; 70: 68–78. - PubMed
-
- Stine JG, Northup PG. Coagulopathy Before and After Liver Transplantation: From the Hepatic to the Systemic Circulatory Systems. Clin Liver Dis 2017; 21: 253–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
