

Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids
- PMID: 30846318
- PMCID: PMC7696670
- DOI: 10.1016/j.molcel.2019.01.036
Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids
Abstract
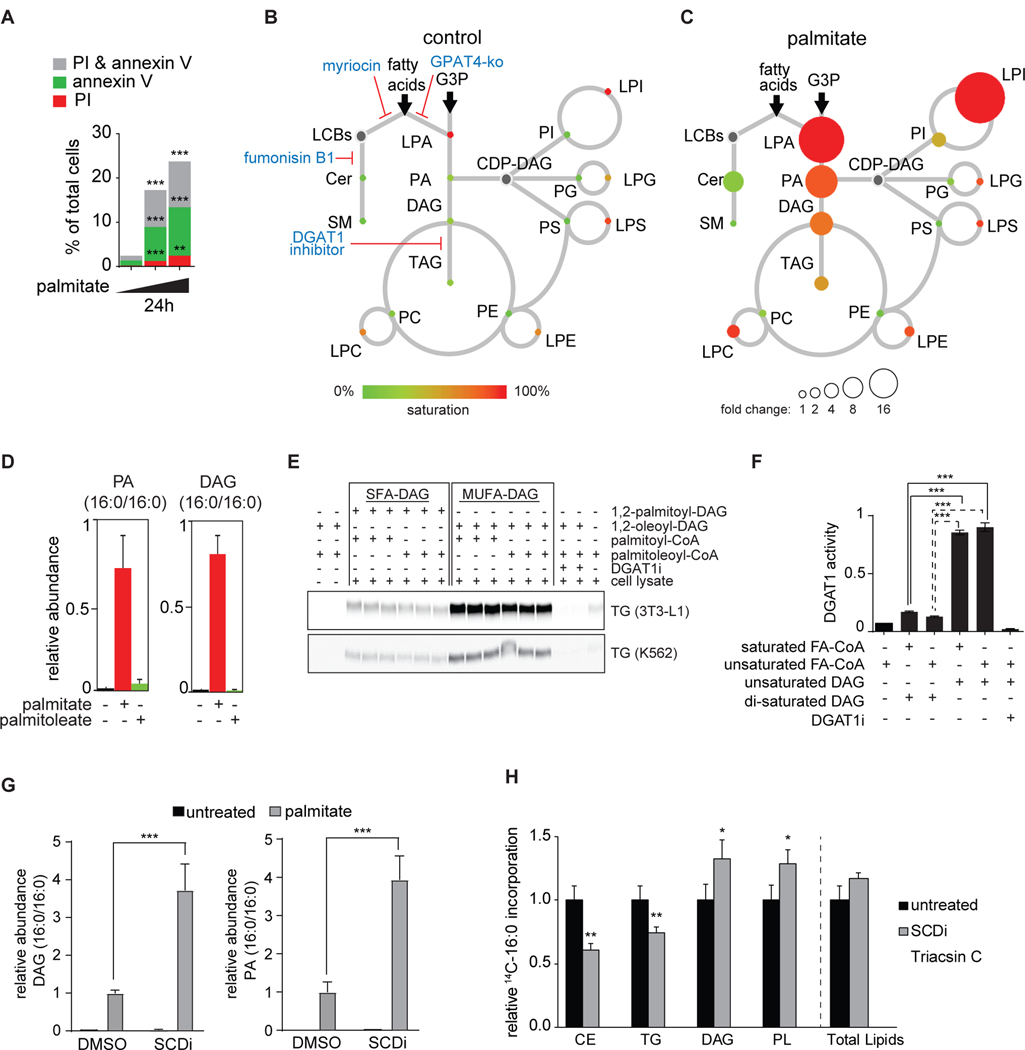
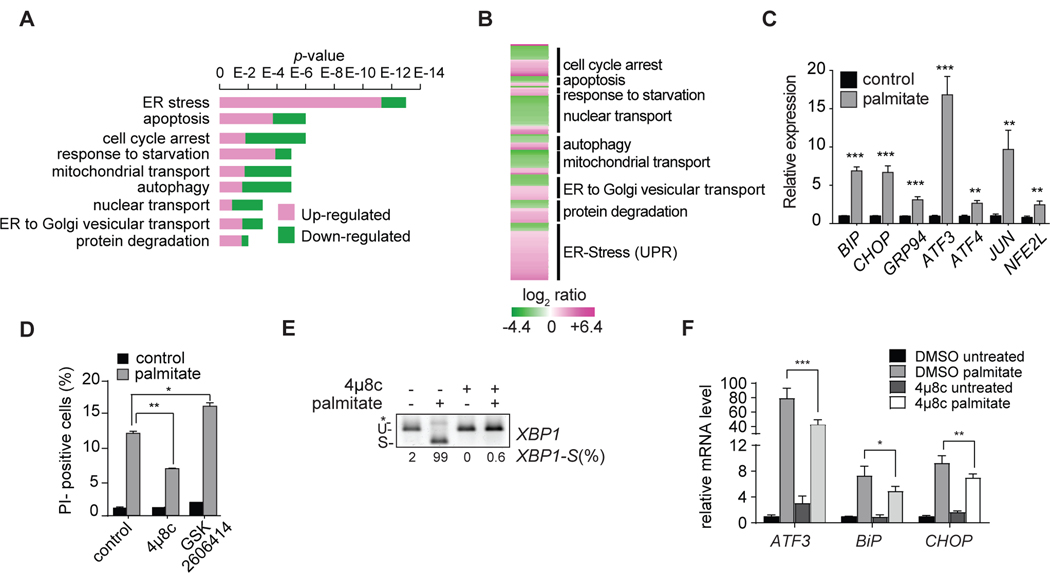
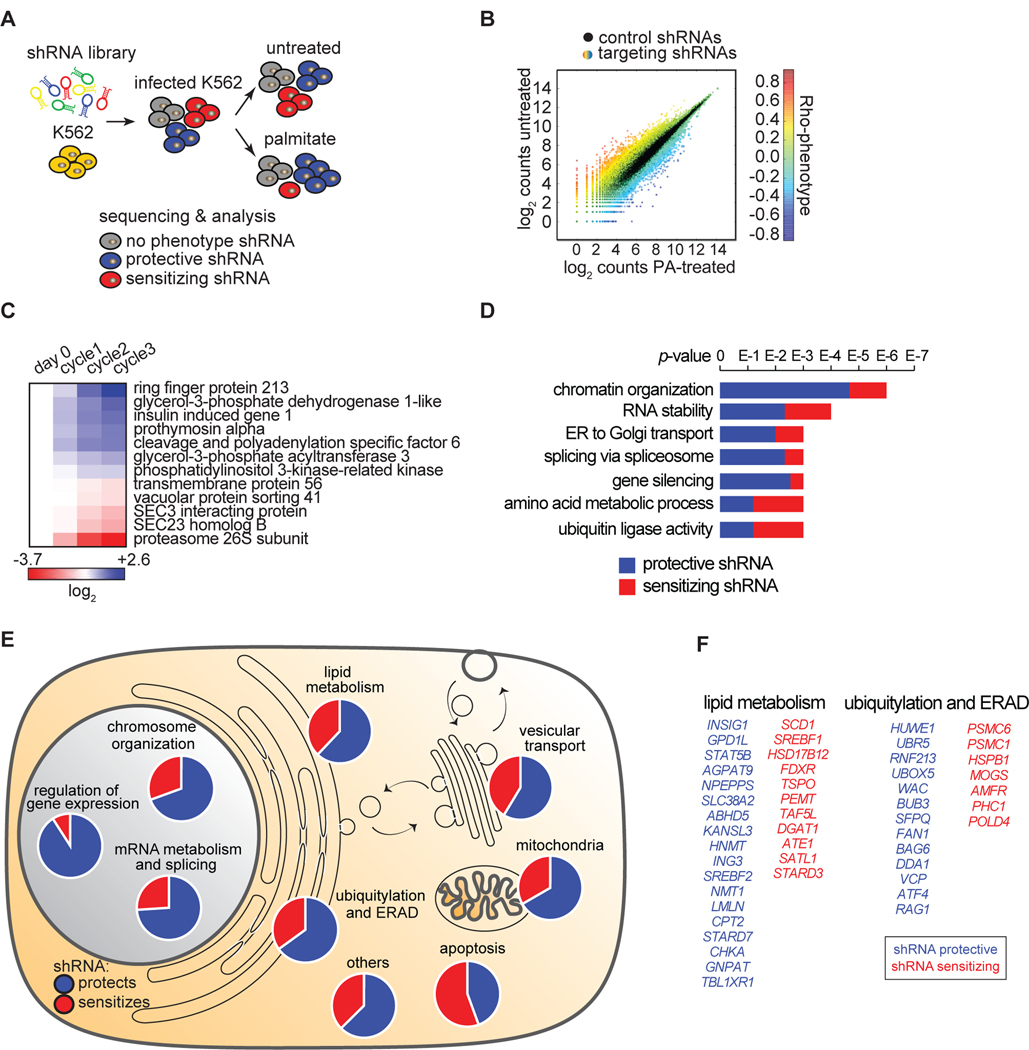
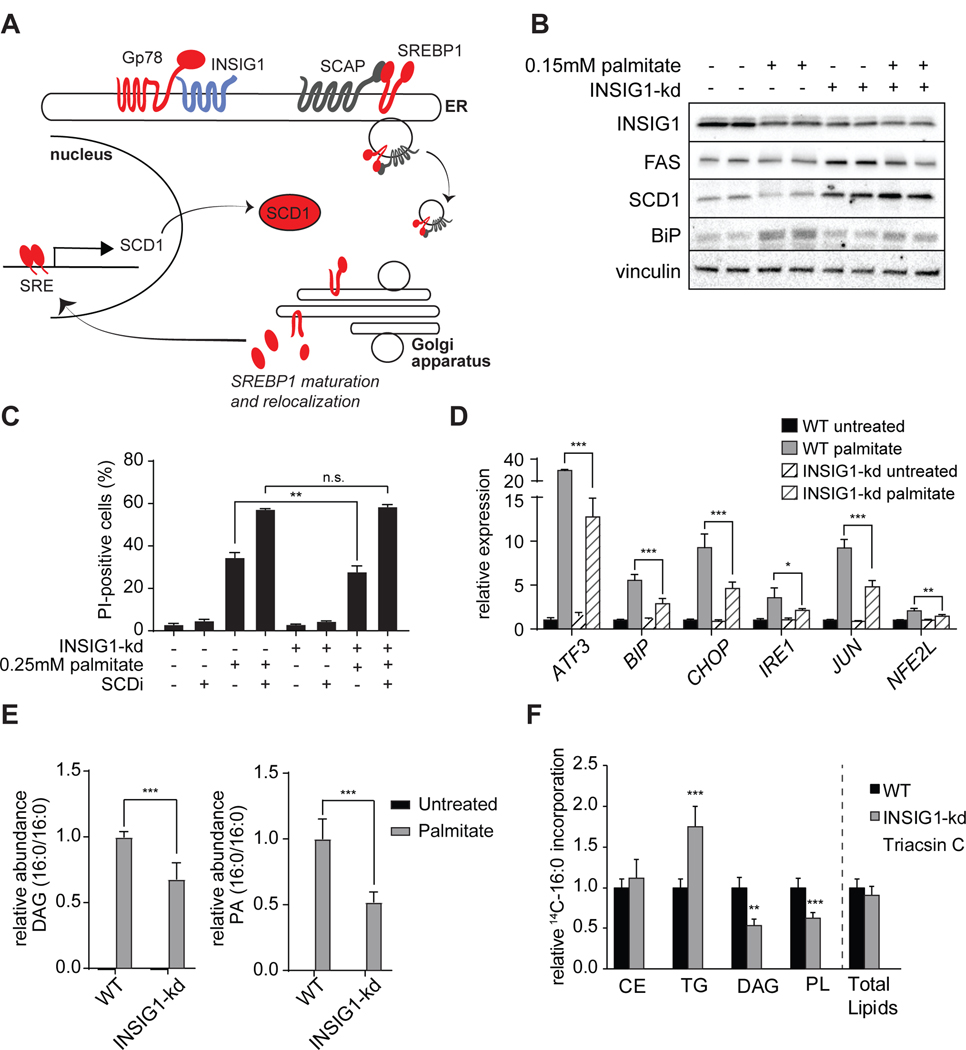
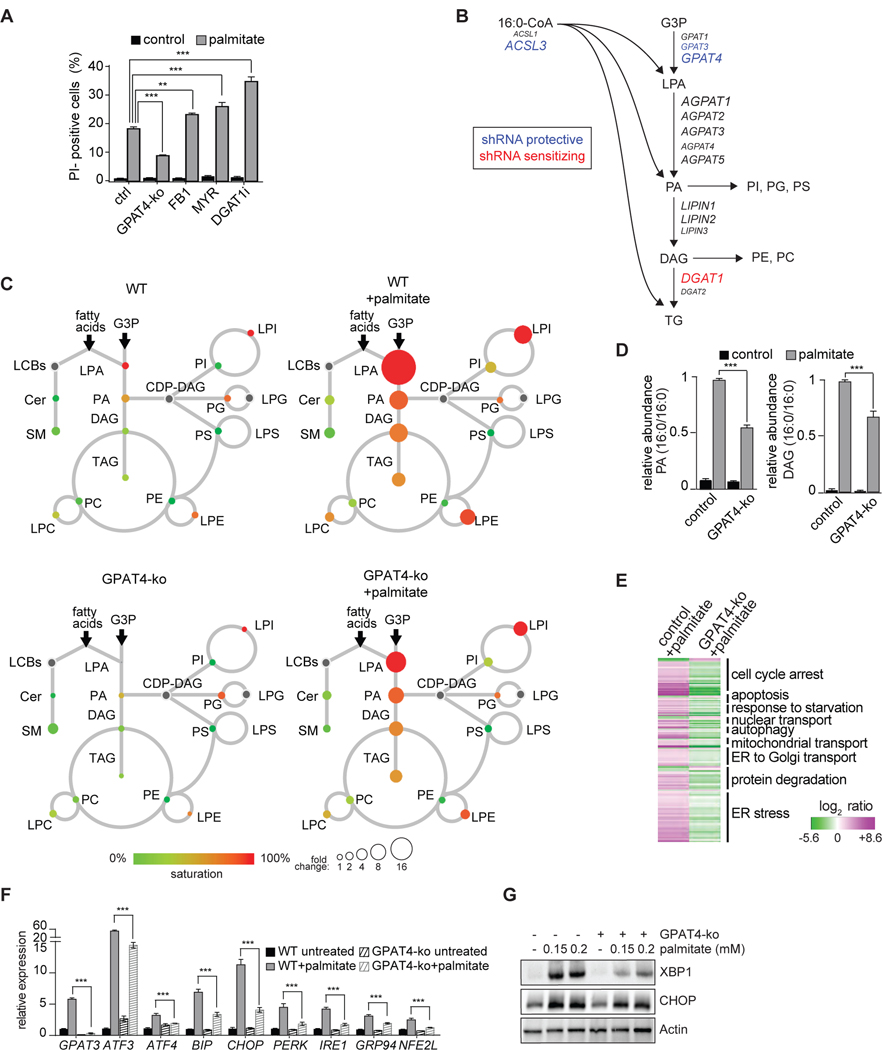
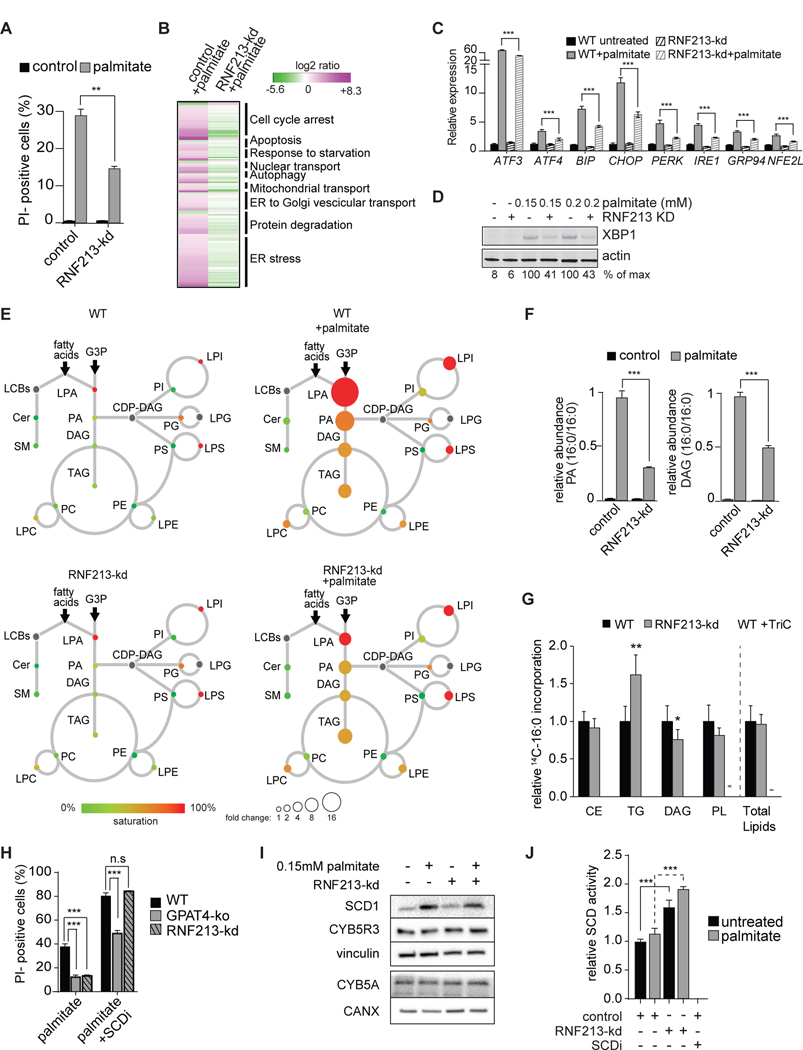
Excessive levels of saturated fatty acids are toxic to cells, although the basis for this lipotoxicity remains incompletely understood. Here, we analyzed the transcriptome, lipidome, and genetic interactions of human leukemia cells exposed to palmitate. Palmitate treatment increased saturated glycerolipids, accompanied by a transcriptional stress response, including upregulation of the endoplasmic reticulum (ER) stress response. A comprehensive genome-wide short hairpin RNA (shRNA) screen identified >350 genes modulating lipotoxicity. Among previously unknown genetic modifiers of lipotoxicity, depletion of RNF213, a putative ubiquitin ligase mutated in Moyamoya vascular disease, protected cells from lipotoxicity. On a broader level, integration of our comprehensive datasets revealed that changes in di-saturated glycerolipids, but not other lipid classes, are central to lipotoxicity in this model. Consistent with this, inhibition of ER-localized glycerol-3-phosphate acyltransferase activity protected from all aspects of lipotoxicity. Identification of genes modulating the response to saturated fatty acids may reveal novel therapeutic strategies for treating metabolic diseases linked to lipotoxicity.
Keywords: Moyamoya disease; glycerolipid; lipotoxicity; palmitate; saturated fatty acid.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
J.W.H. is a consultant and founder of Rheostat Therapeutics and a consultant for X-Chem Inc.
Figures
References
-
- Agarwal AK, Sukumaran S, Cortes VA, Tunison K, Mizrachi D, Sankella S, Gerard RD, Horton JD, and Garg A. (2011). Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(−/−) gene lipodystrophic mice. J Biol Chem 286, 37676–37691. - PMC - PubMed
-
- Beausoleil SA, Villen J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24, 1285–1292. - PubMed
-
- Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, and Schaffer JE (2006). Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47, 2726–2737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
