

Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities
- PMID: 30846875
- PMCID: PMC6727648
- DOI: 10.1038/s41569-019-0169-2
Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities
Abstract
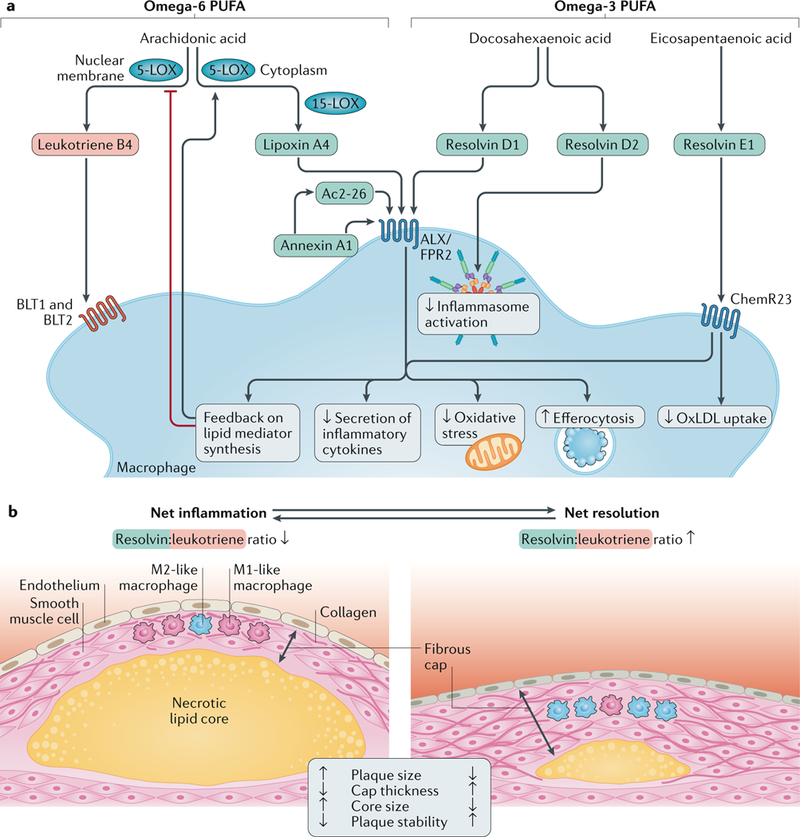
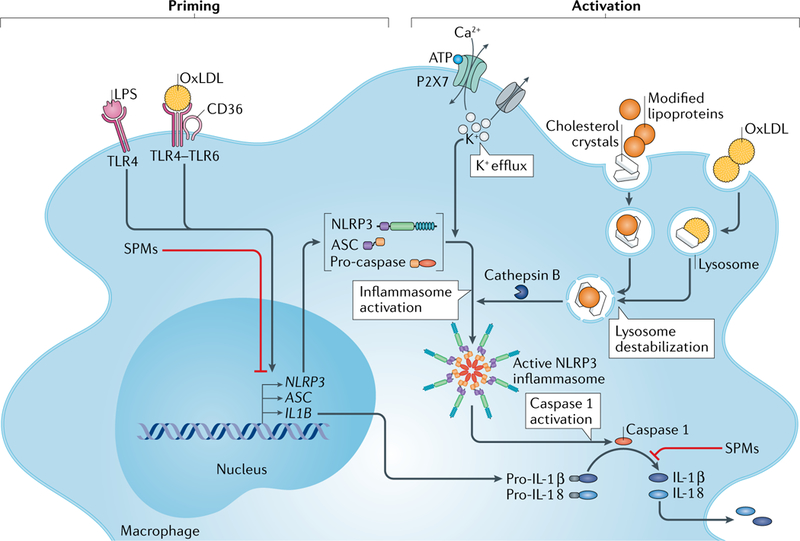
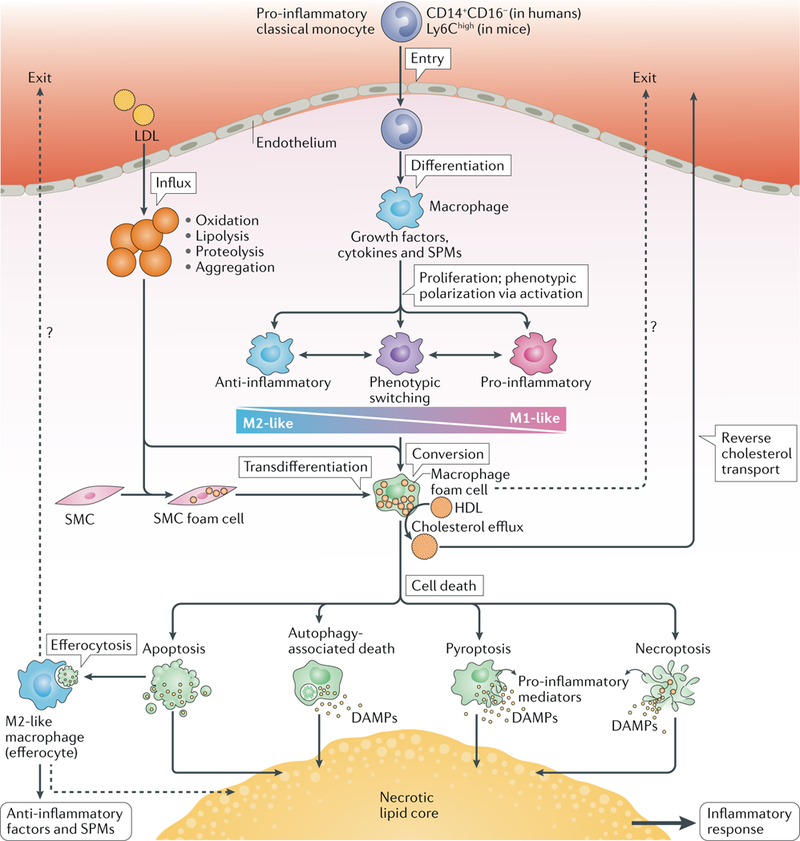
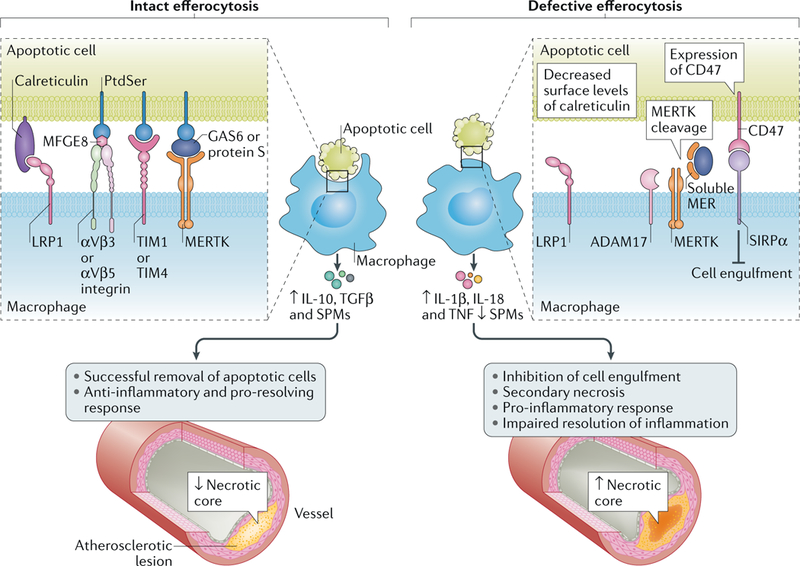
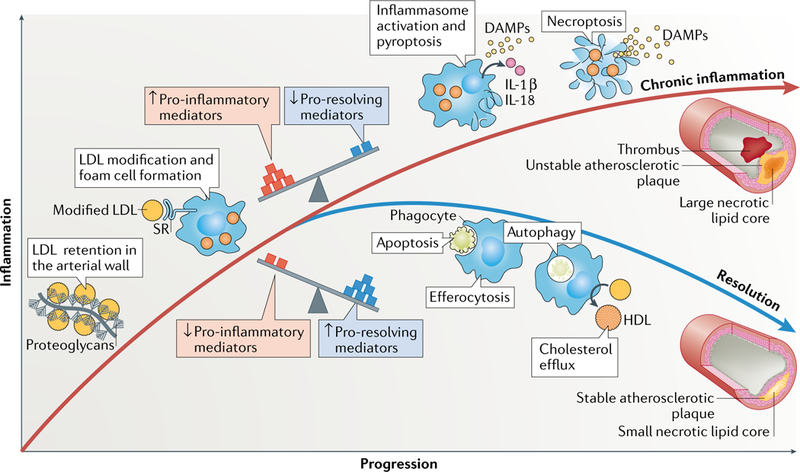
Atherosclerosis is a lipid-driven inflammatory disease of the arterial intima in which the balance of pro-inflammatory and inflammation-resolving mechanisms dictates the final clinical outcome. Intimal infiltration and modification of plasma-derived lipoproteins and their uptake mainly by macrophages, with ensuing formation of lipid-filled foam cells, initiate atherosclerotic lesion formation, and deficient efferocytotic removal of apoptotic cells and foam cells sustains lesion progression. Defective efferocytosis, as a sign of inadequate inflammation resolution, leads to accumulation of secondarily necrotic macrophages and foam cells and the formation of an advanced lesion with a necrotic lipid core, indicative of plaque vulnerability. Resolution of inflammation is mediated by specialized pro-resolving lipid mediators derived from omega-3 fatty acids or arachidonic acid and by relevant proteins and signalling gaseous molecules. One of the major effects of inflammation resolution mediators is phenotypic conversion of pro-inflammatory macrophages into macrophages that suppress inflammation and promote healing. In advanced atherosclerotic lesions, the ratio between specialized pro-resolving mediators and pro-inflammatory lipids (in particular leukotrienes) is strikingly low, providing a molecular explanation for the defective inflammation resolution features of these lesions. In this Review, we discuss the mechanisms of the formation of clinically dangerous atherosclerotic lesions and the potential of pro-resolving mediator therapy to inhibit this process.
Figures
References
-
- Williams KJ & Tabas I The response-to-retention hypothesis of atherogenesis reinforced. Curr. Opin. Lipidol. 9, 471–474 (1998). - PubMed
-
- Tabas I, Williams KJ & Boren J Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116, 1832–1844 (2007). - PubMed
-
- Hansson GK Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
