

Large-Scale Comparative Analysis of Codon Models Accounting for Protein and Nucleotide Selection
- PMID: 30847475
- PMCID: PMC6526913
- DOI: 10.1093/molbev/msz048
Large-Scale Comparative Analysis of Codon Models Accounting for Protein and Nucleotide Selection
Abstract
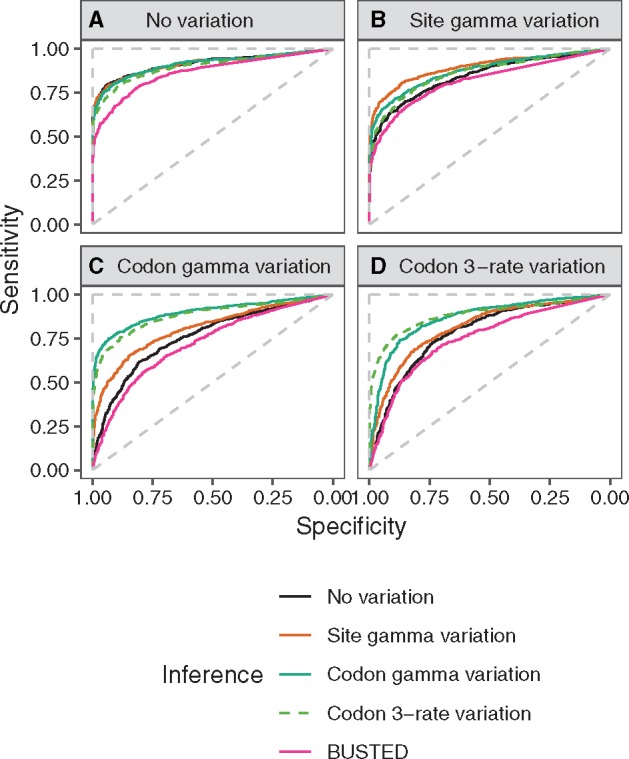
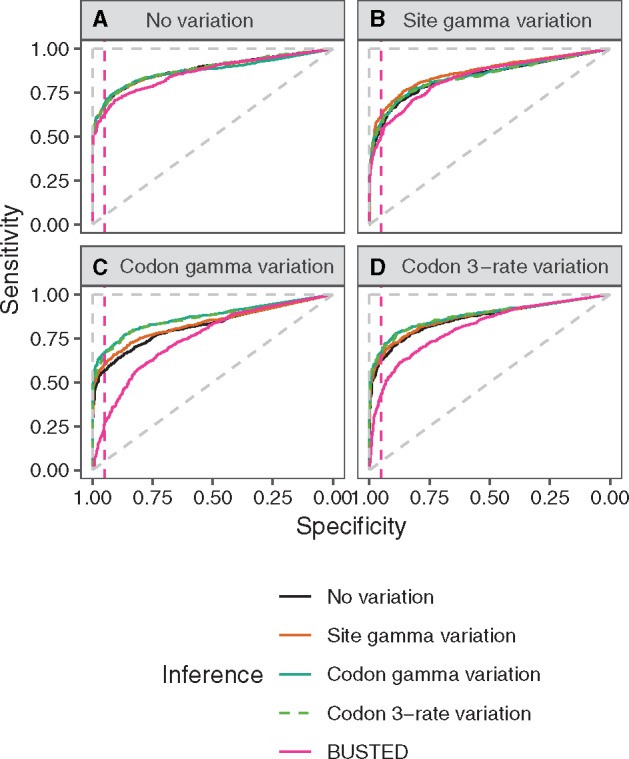
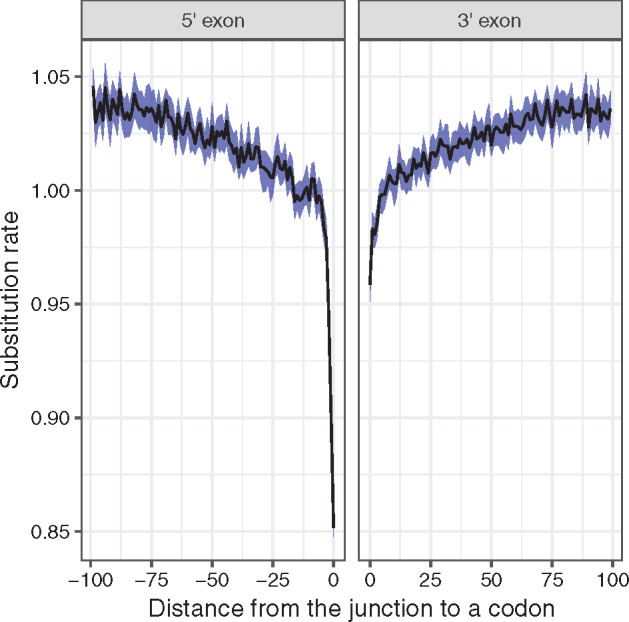
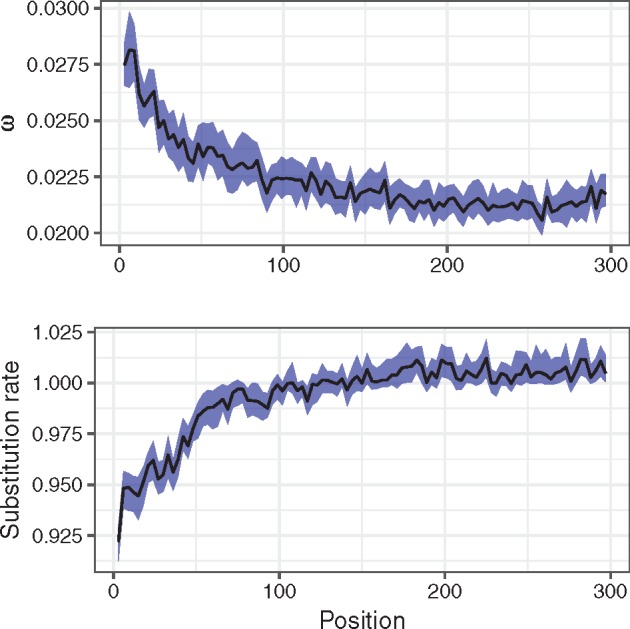
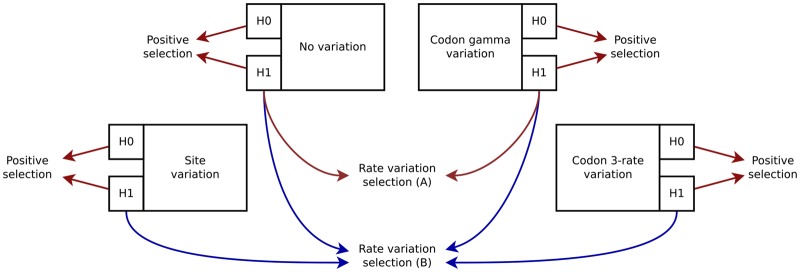
There are numerous sources of variation in the rate of synonymous substitutions inside genes, such as direct selection on the nucleotide sequence, or mutation rate variation. Yet scans for positive selection rely on codon models which incorporate an assumption of effectively neutral synonymous substitution rate, constant between sites of each gene. Here we perform a large-scale comparison of approaches which incorporate codon substitution rate variation and propose our own simple yet effective modification of existing models. We find strong effects of substitution rate variation on positive selection inference. More than 70% of the genes detected by the classical branch-site model are presumably false positives caused by the incorrect assumption of uniform synonymous substitution rate. We propose a new model which is strongly favored by the data while remaining computationally tractable. With the new model we can capture signatures of nucleotide level selection acting on translation initiation and on splicing sites within the coding region. Finally, we show that rate variation is highest in the highly recombining regions, and we propose that recombination and mutation rate variation, such as high CpG mutation rate, are the two main sources of nucleotide rate variation. Although we detect fewer genes under positive selection in Drosophila than without rate variation, the genes which we detect contain a stronger signal of adaptation of dynein, which could be associated with Wolbachia infection. We provide software to perform positive selection analysis using the new model.
Keywords: codon models; positive selection; substitution rate variation; synonymous substitutions.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Alexa A, Rahnenfuhrer J.. 2016. topGO: enrichment analysis for gene ontology. R Package Version 2(28.0).
-
- Baele G, Lemey P.. 2013. Bayesian evolutionary model testing in the phylogenomics era: matching model complexity with computational efficiency. Bioinformatics 2916: 1970–1979. - PubMed
-
- Betancur-R R, Orti G, Pyron RA.. 2015. Fossil-based comparative analyses reveal ancient marine ancestry erased by extinction in ray-finned fishes. Ecol Lett. 185: 441–450. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
