

Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: the role of integrated systems
- PMID: 30848350
- PMCID: PMC6476637
- DOI: 10.1007/s00424-019-02269-0
Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: the role of integrated systems
Abstract
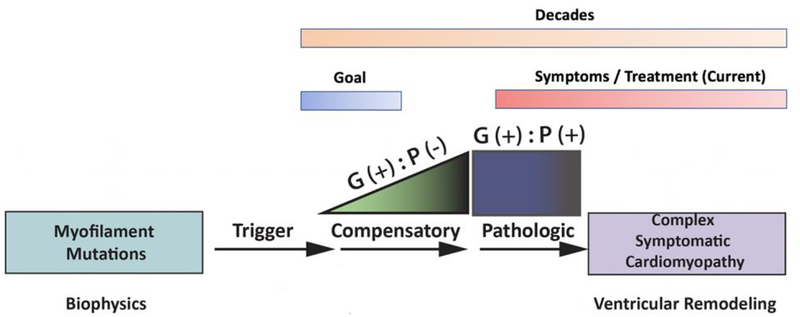
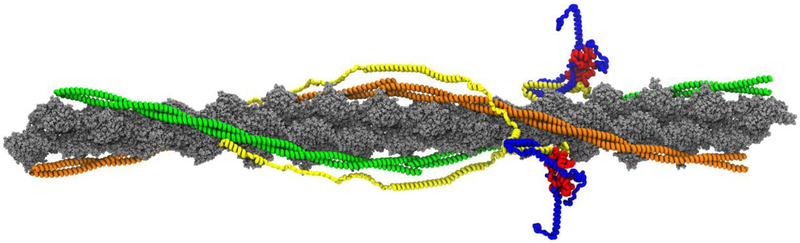
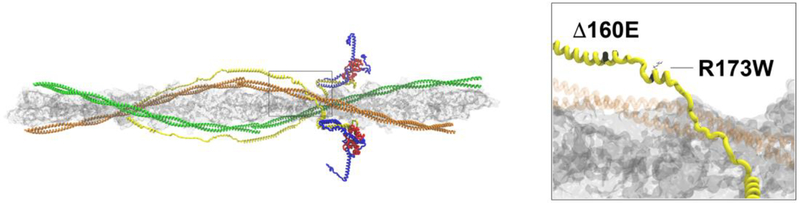
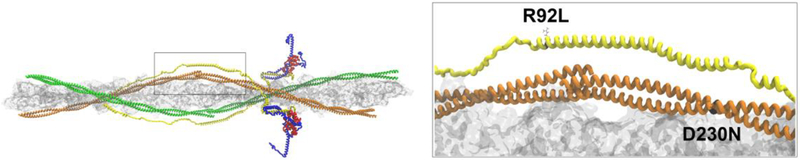
The classic clinical definition of hypertrophic cardiomyopathy (HCM) as originally described by Teare is deceptively simple, "left ventricular hypertrophy in the absence of any identifiable cause." Longitudinal studies, however, including a seminal study performed by Frank and Braunwald in 1968, clearly described the disorder much as we know it today, a complex, progressive, and highly variable cardiomyopathy affecting ~ 1/500 individuals worldwide. Subsequent genetic linkage studies in the early 1990s identified mutations in virtually all of the protein components of the cardiac sarcomere as the primary molecular cause of HCM. In addition, a substantial proportion of inherited dilated cardiomyopathy (DCM) has also been linked to sarcomeric protein mutations. Despite our deep understanding of the overall function of the sarcomere as the primary driver of cardiac contractility, the ability to use genotype in patient management remains elusive. A persistent challenge in the field from both the biophysical and clinical standpoints is how to rigorously link high-resolution protein dynamics and mechanics to the long-term cardiovascular remodeling process that characterizes these complex disorders. In this review, we will explore the depth of the problem from both the standpoint of a multi-subunit, highly conserved and dynamic "machine" to the resultant clinical and structural human phenotype with an emphasis on new, integrative approaches that can be widely applied to identify both novel disease mechanisms and new therapeutic targets for these primary biophysical disorders of the cardiac sarcomere.
Keywords: Dilated cardiomyopathy; Hypertrophic cardiomyopathy; Thin filament; Tropomyosin; Troponin.
Figures
References
-
- Axelsson A, Iversen K, Vejlstrup N, Ho C, Norsk J, Langhoff L, Ahtarovski K, Corell P, Havndrup O, Jensen M, Bundgaard H (2015) Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. The Lancet Diabetes & Endocrinology 3:123–131. doi:10.1016/S2213-8587(14)70241-4 - DOI - PubMed
-
- Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, Carniel E, Fain PR, Aragona P, Di Lenarda A, Mestroni L, Taylor MRG (2013) Whole Exome Sequencing Identifies a Troponin T Mutation Hot Spot in Familial Dilated Cardiomyopathy. PLoS ONE 8:e78104. doi:10.1371/journal.pone.0078104 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
