

Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis
- PMID: 30849372
- PMCID: PMC6424819
- DOI: 10.1016/j.cell.2019.02.012
Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis
Abstract
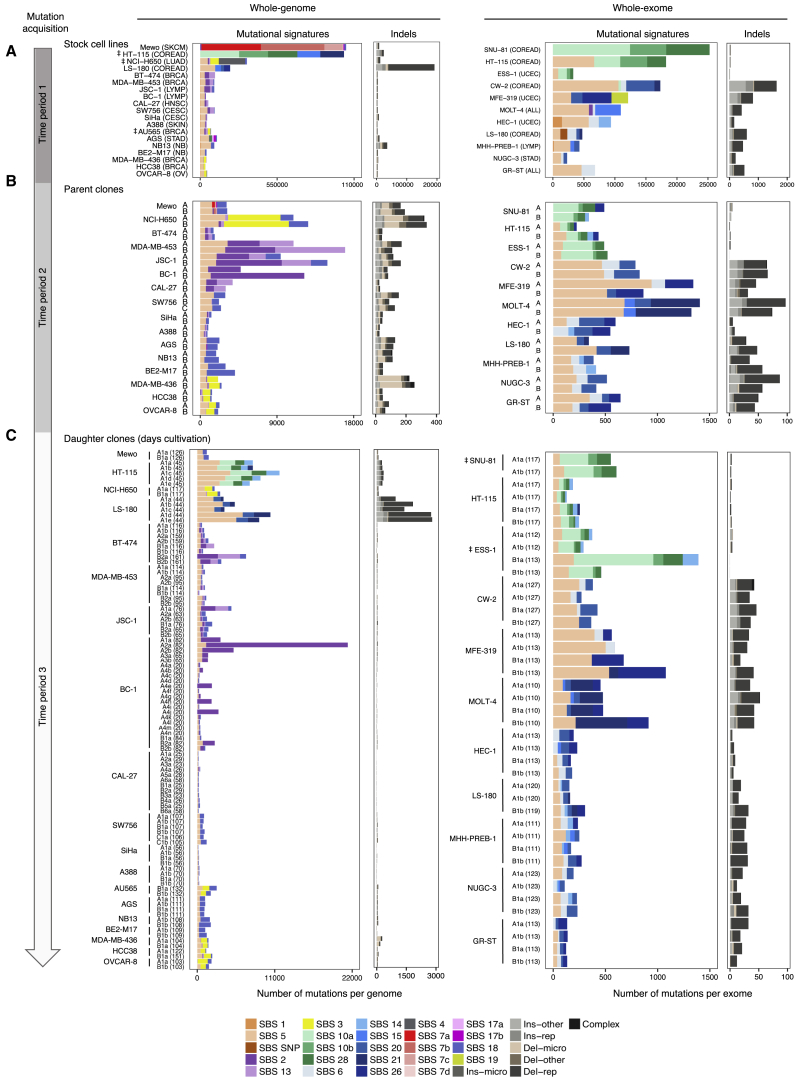
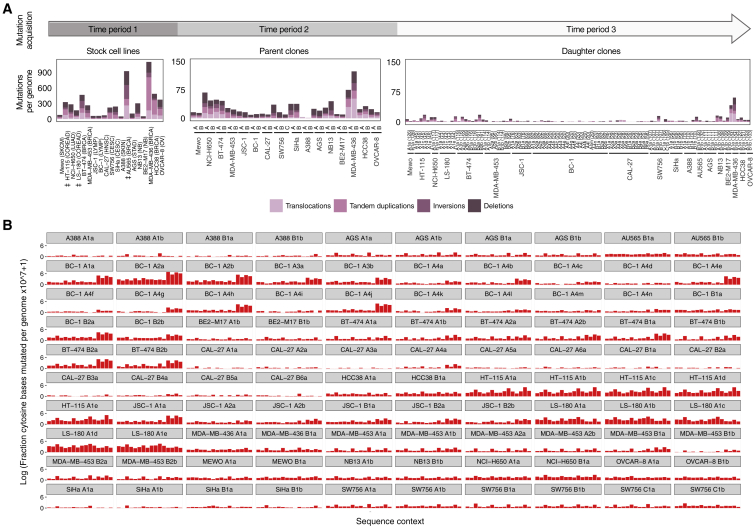
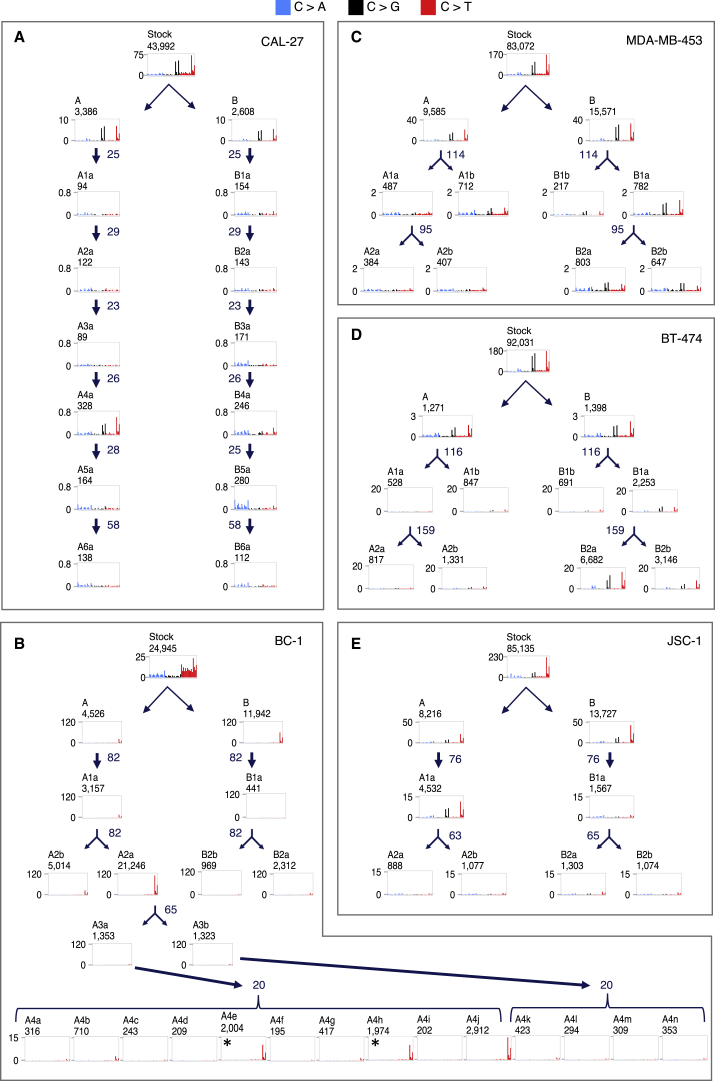
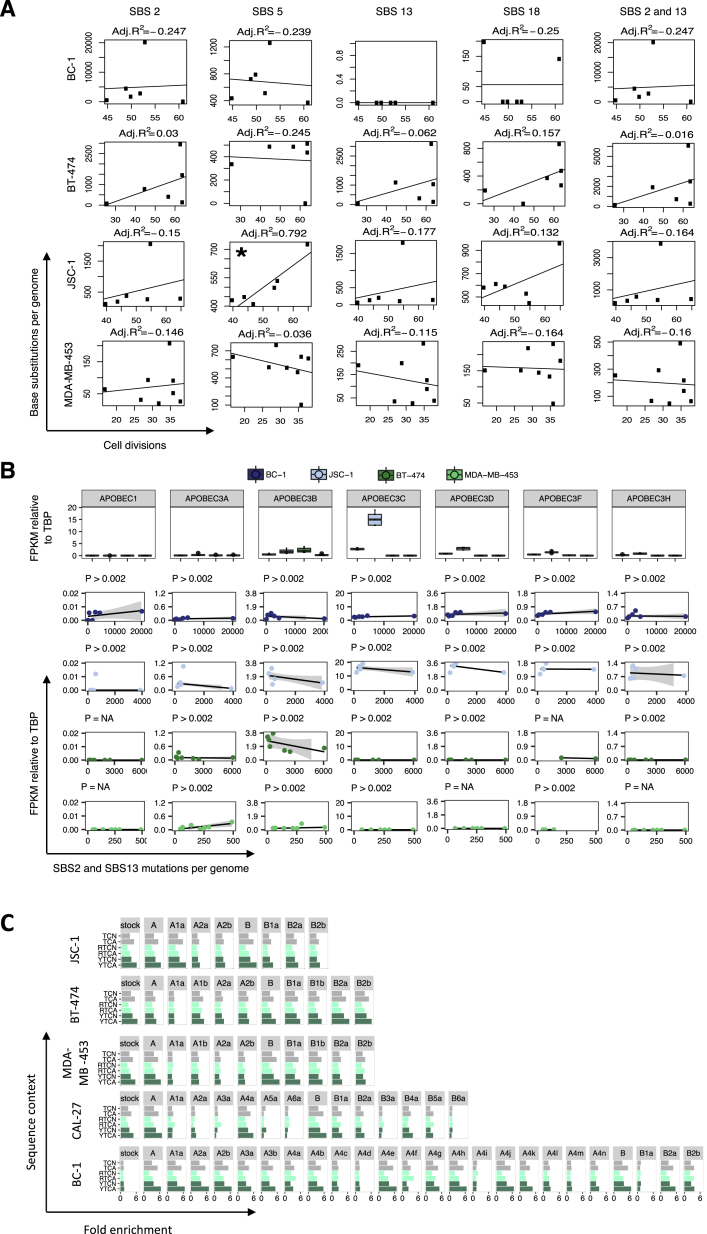
Multiple signatures of somatic mutations have been identified in cancer genomes. Exome sequences of 1,001 human cancer cell lines and 577 xenografts revealed most common mutational signatures, indicating past activity of the underlying processes, usually in appropriate cancer types. To investigate ongoing patterns of mutational-signature generation, cell lines were cultured for extended periods and subsequently DNA sequenced. Signatures of discontinued exposures, including tobacco smoke and ultraviolet light, were not generated in vitro. Signatures of normal and defective DNA repair and replication continued to be generated at roughly stable mutation rates. Signatures of APOBEC cytidine deaminase DNA-editing exhibited substantial fluctuations in mutation rate over time with episodic bursts of mutations. The initiating factors for the bursts are unclear, although retrotransposon mobilization may contribute. The examined cell lines constitute a resource of live experimental models of mutational processes, which potentially retain patterns of activity and regulation operative in primary human cancers.
Keywords: APOBEC deaminases; cancer cell lines; episodic mutagenesis; mutational signatures; xenografts.
Crown Copyright © 2019. Published by Elsevier Inc. All rights reserved.
Figures
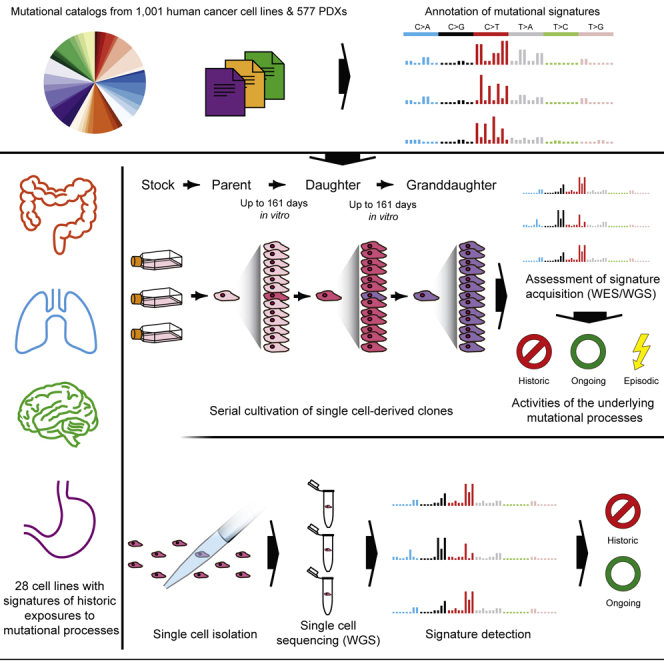
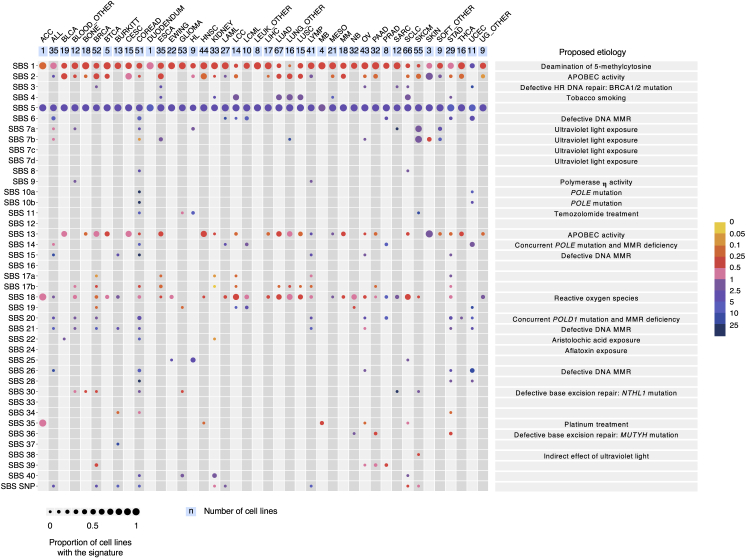
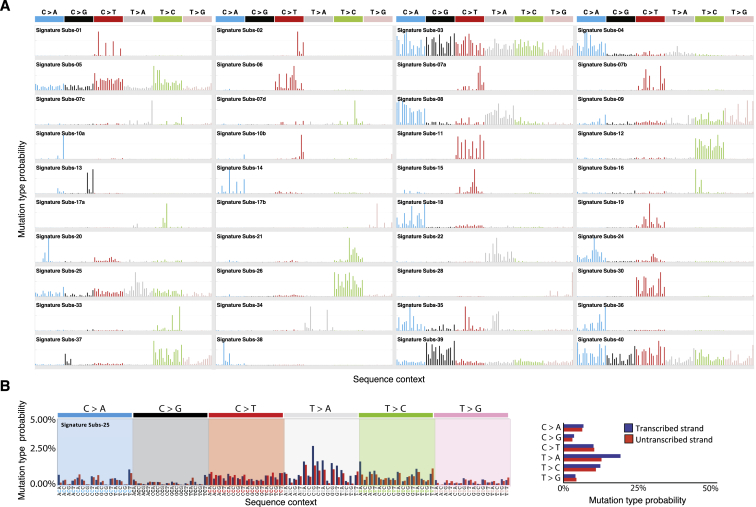
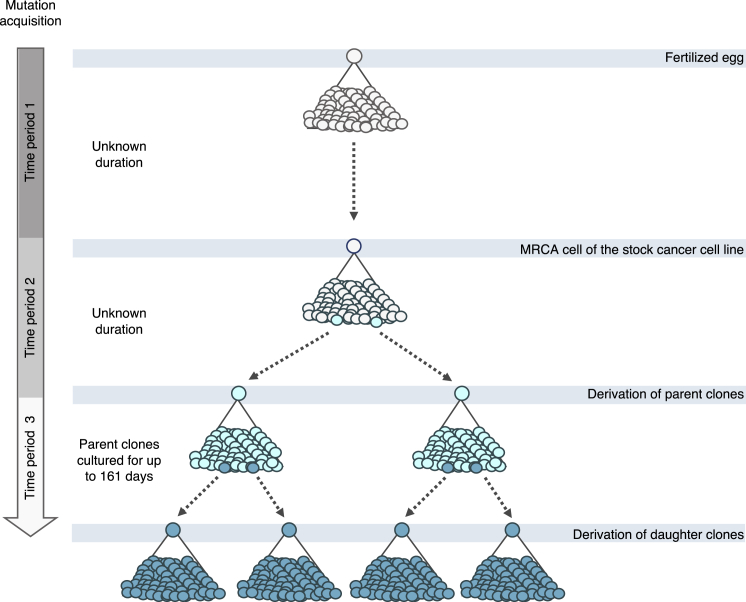

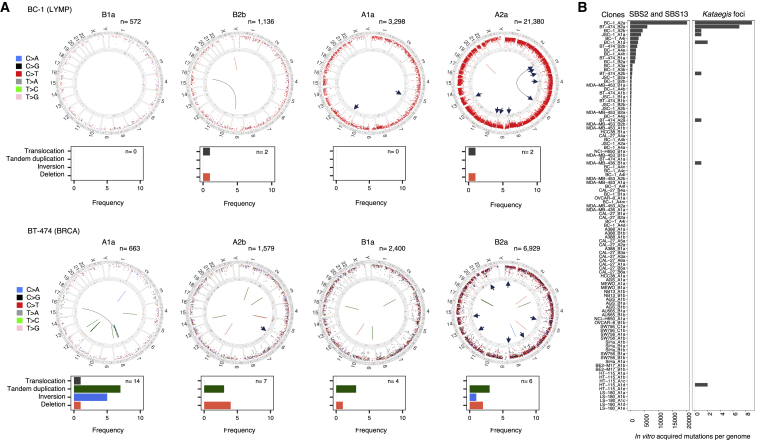
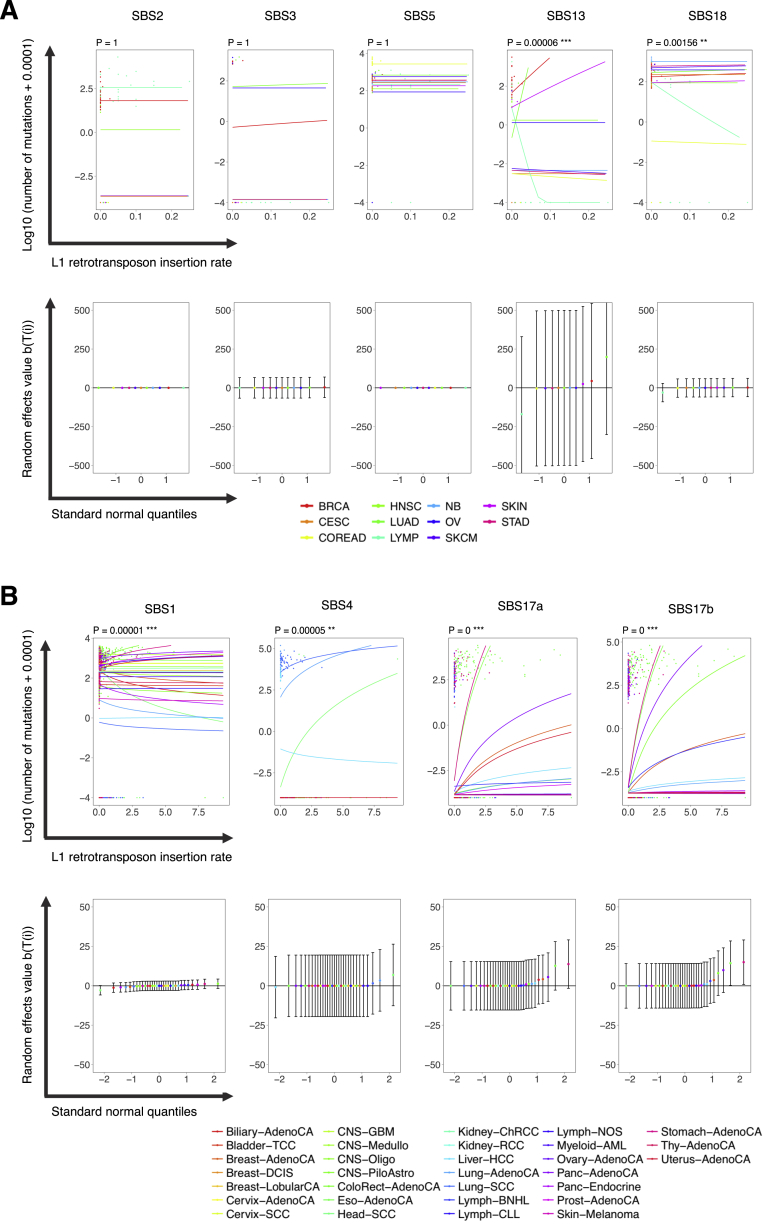
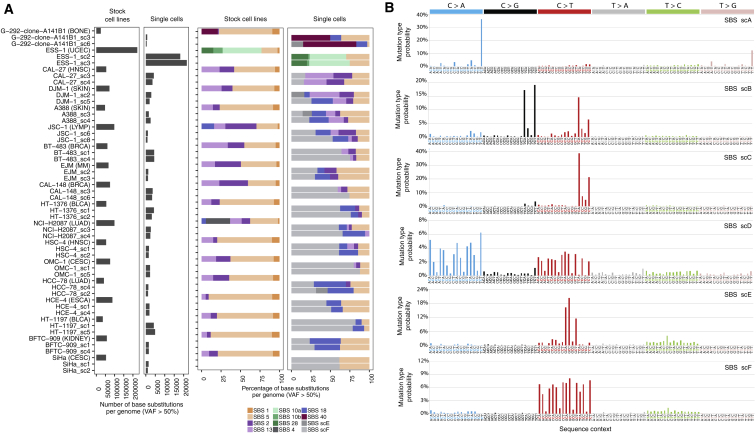
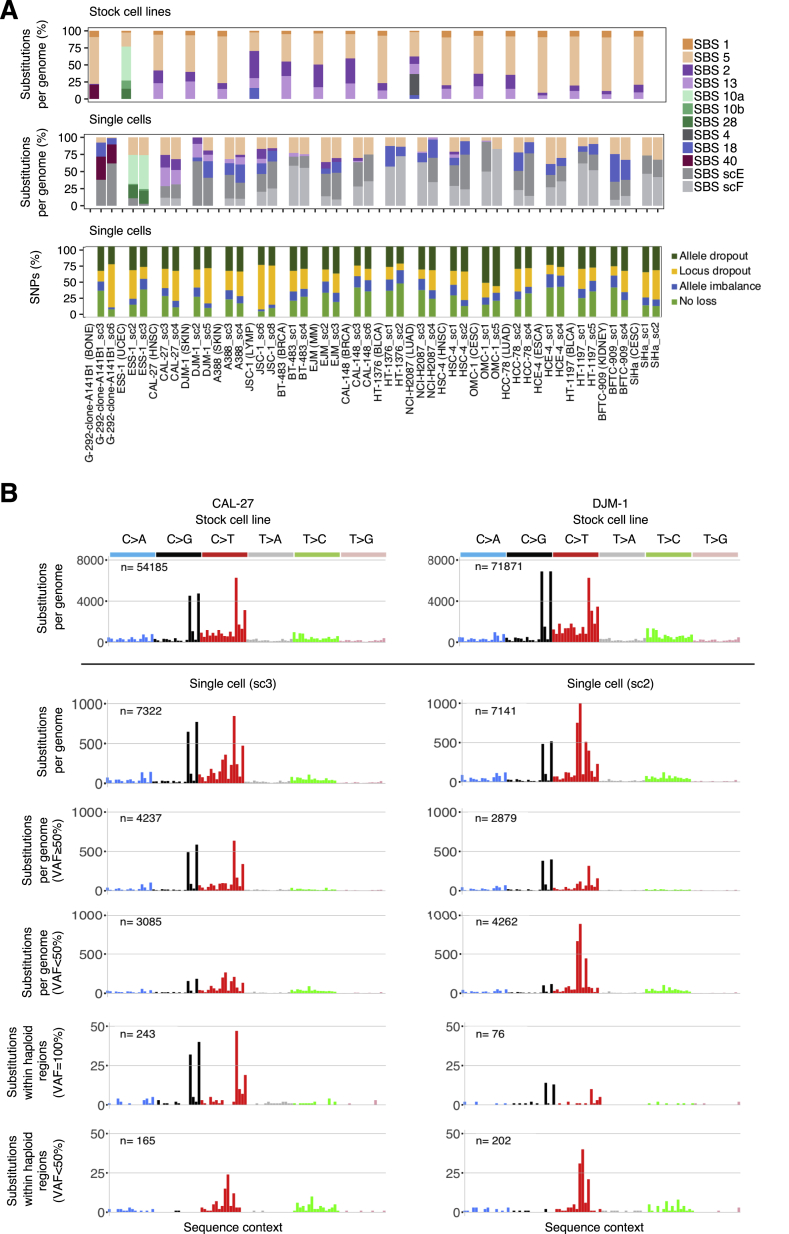
Comment in
-
Switching APOBEC mutation signatures.Nat Rev Genet. 2019 May;20(5):253. doi: 10.1038/s41576-019-0116-4. Nat Rev Genet. 2019. PMID: 30890789 No abstract available.
References
-
- Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Ng A.W.T., Boot A., Covington K.R., Gordenin D.A., Bergstrom E., Lopez-Bigas N. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv. 2018
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
