

Lipoprotein lipase is active as a monomer
- PMID: 30850549
- PMCID: PMC6442593
- DOI: 10.1073/pnas.1900983116
Lipoprotein lipase is active as a monomer
Abstract
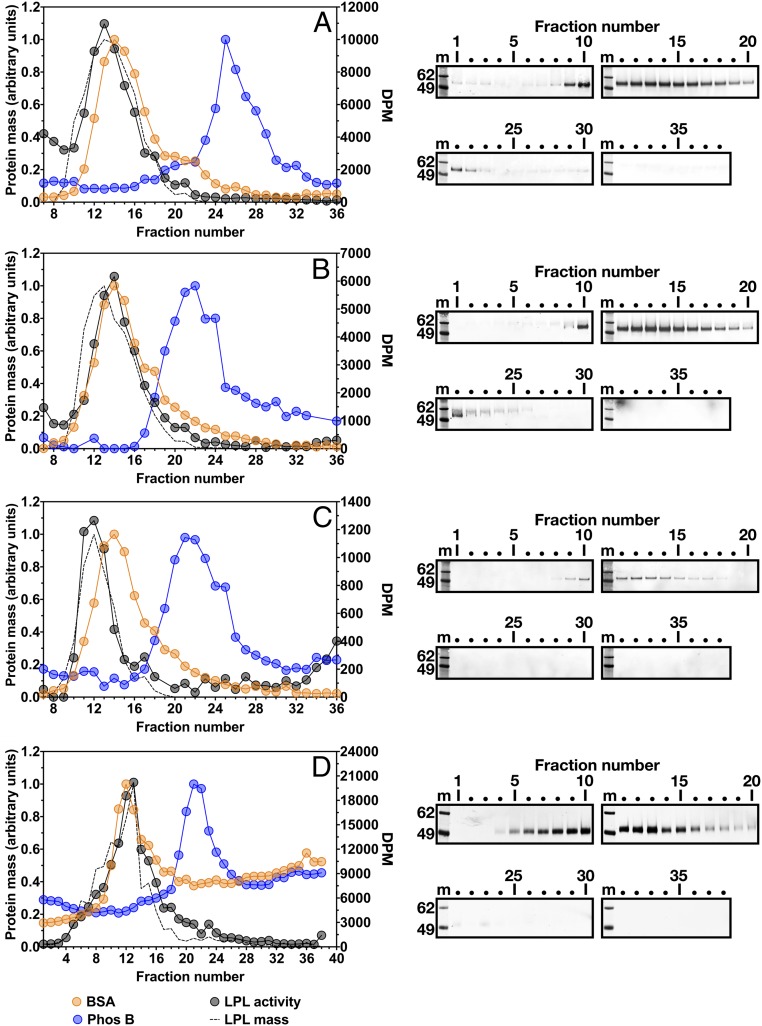
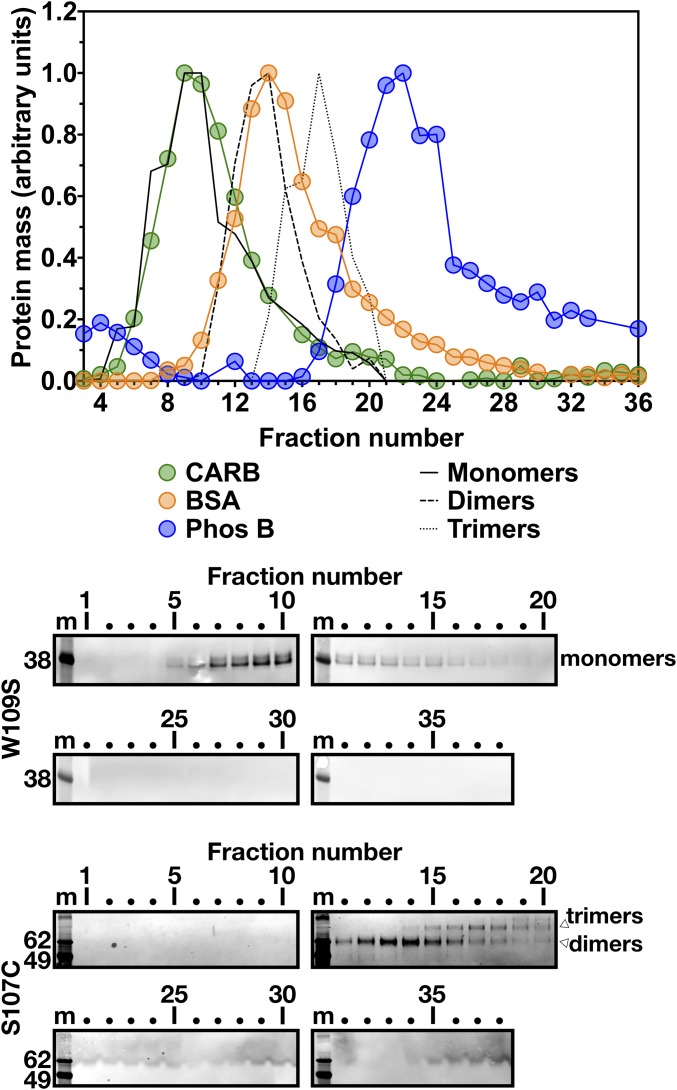
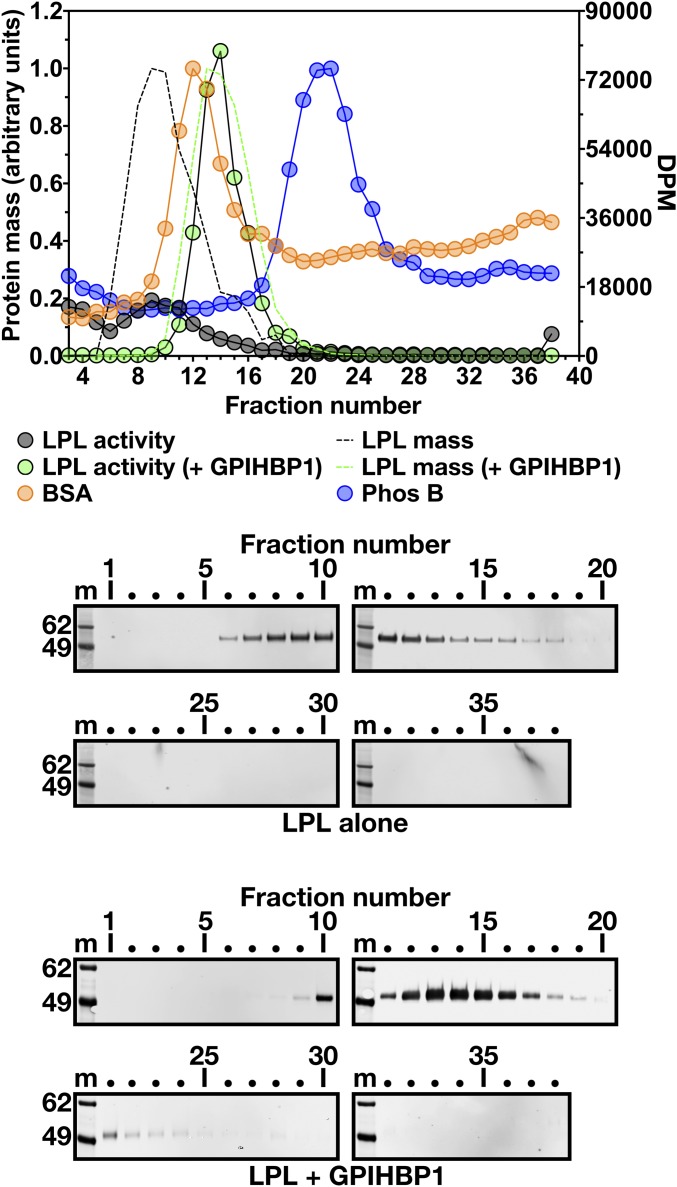
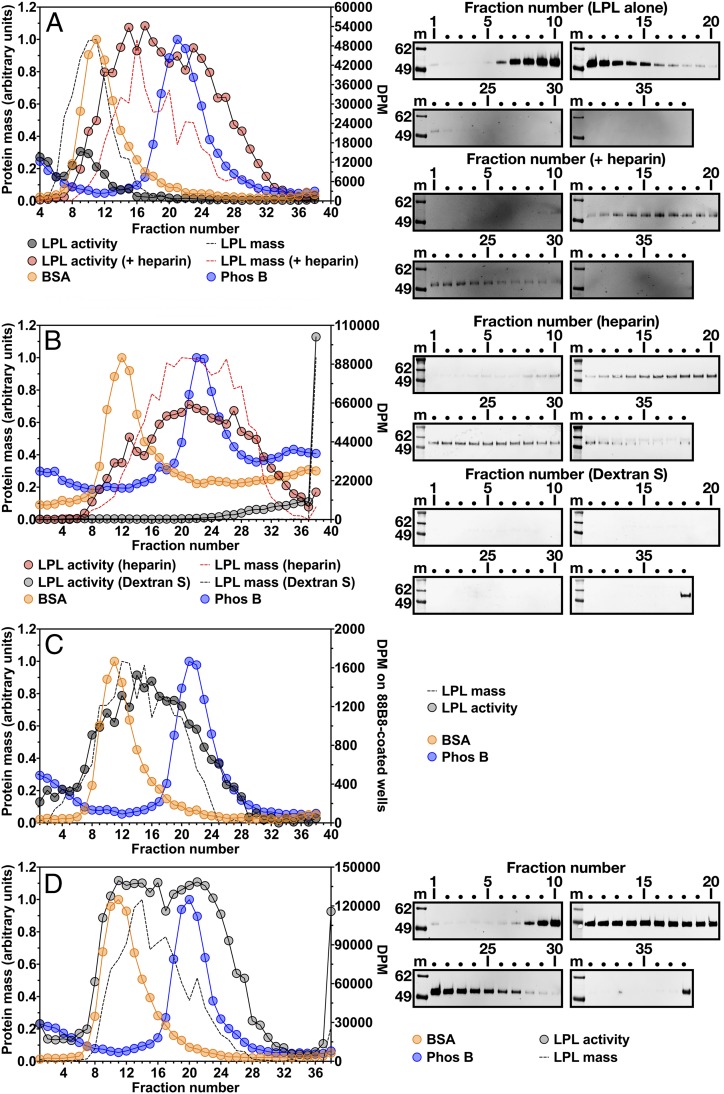
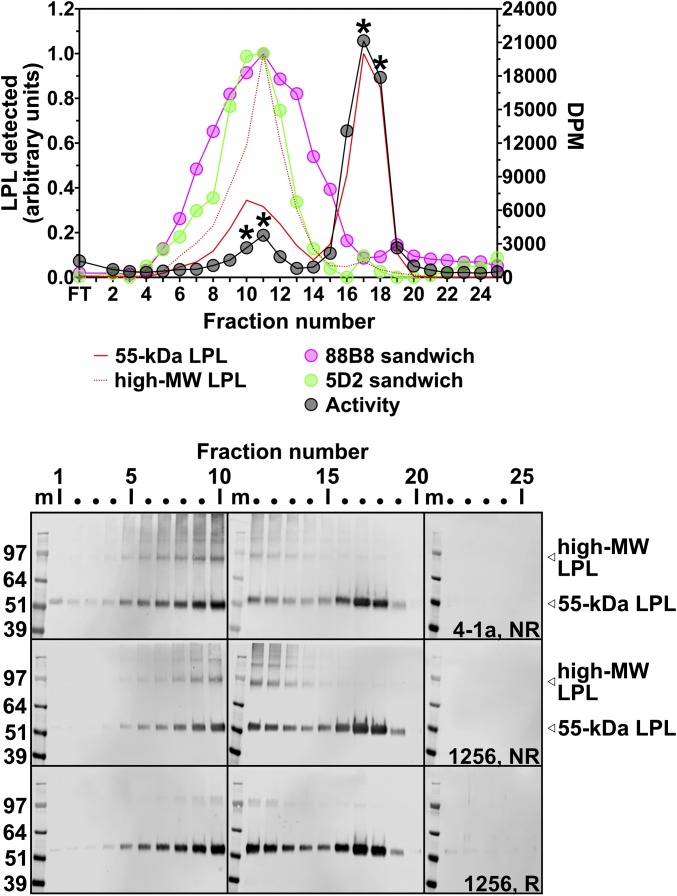
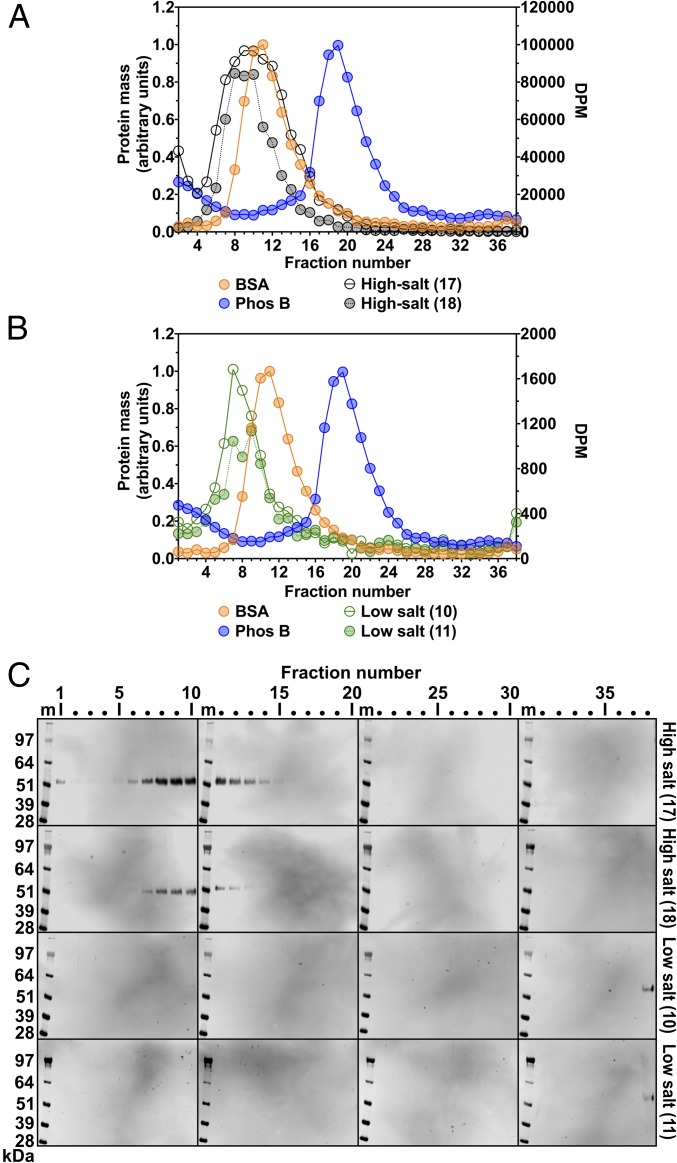
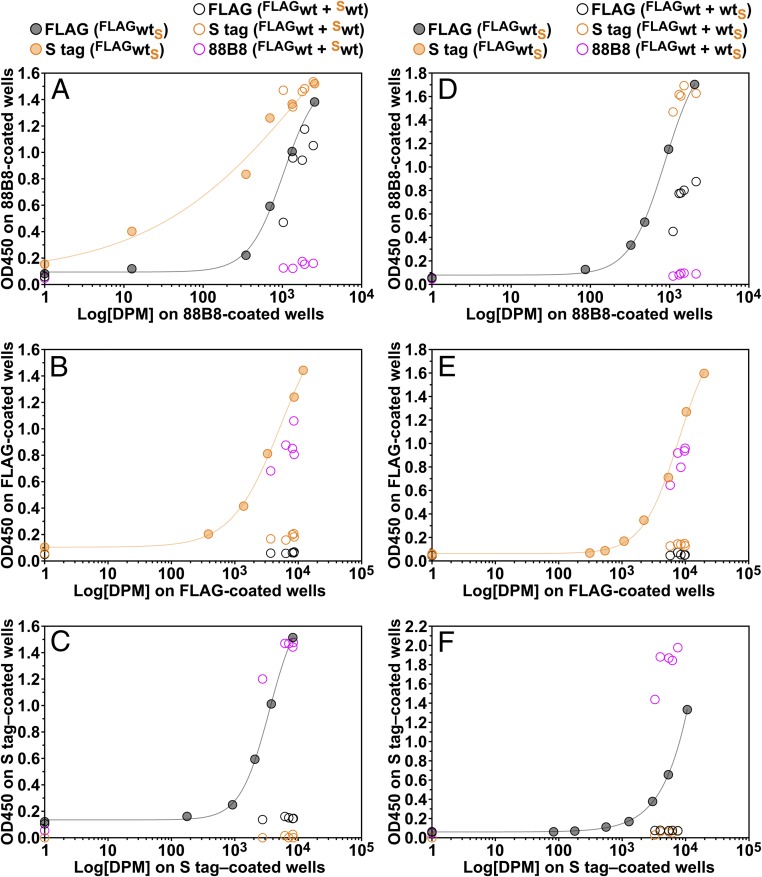
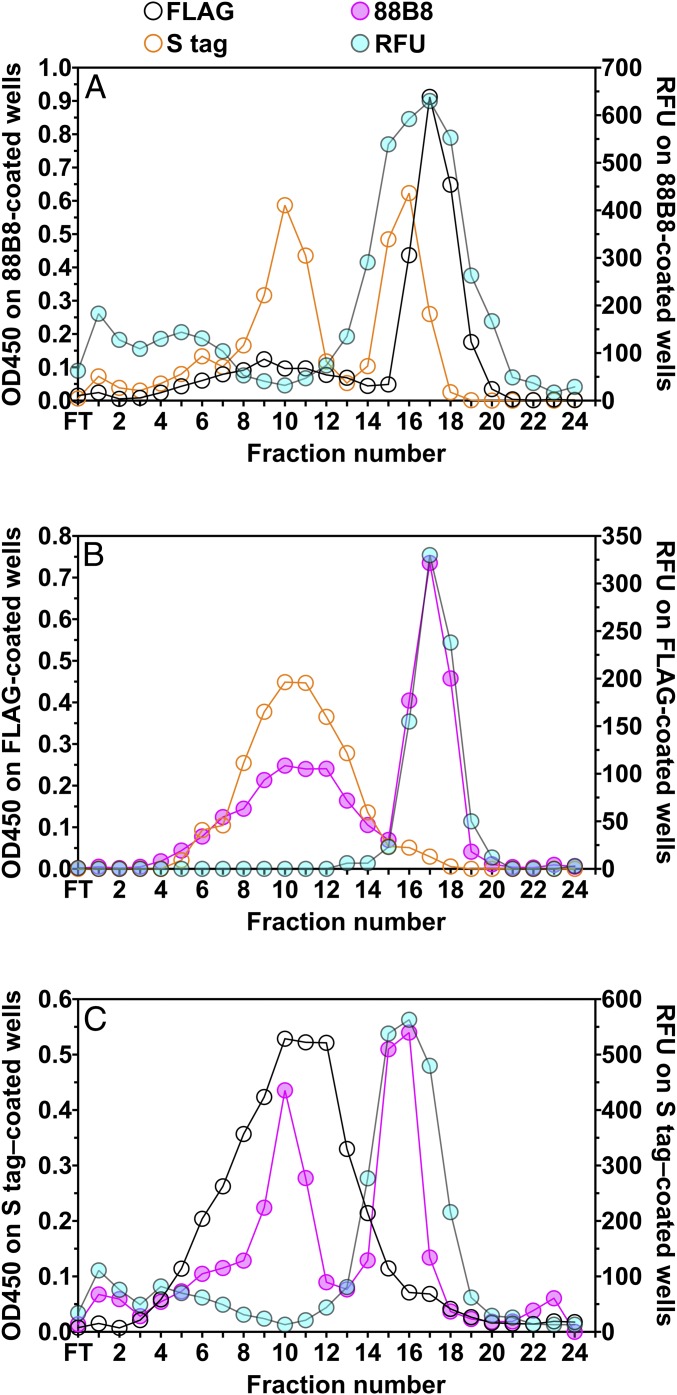
Lipoprotein lipase (LPL), the enzyme that hydrolyzes triglycerides in plasma lipoproteins, is assumed to be active only as a homodimer. In support of this idea, several groups have reported that the size of LPL, as measured by density gradient ultracentrifugation, is ∼110 kDa, twice the size of LPL monomers (∼55 kDa). Of note, however, in those studies the LPL had been incubated with heparin, a polyanionic substance that binds and stabilizes LPL. Here we revisited the assumption that LPL is active only as a homodimer. When freshly secreted human LPL (or purified preparations of LPL) was subjected to density gradient ultracentrifugation (in the absence of heparin), LPL mass and activity peaks exhibited the size expected of monomers (near the 66-kDa albumin standard). GPIHBP1-bound LPL also exhibited the size expected for a monomer. In the presence of heparin, LPL size increased, overlapping with a 97.2-kDa standard. We also used density gradient ultracentrifugation to characterize the LPL within the high-salt and low-salt peaks from a heparin-Sepharose column. The catalytically active LPL within the high-salt peak exhibited the size of monomers, whereas most of the inactive LPL in the low-salt peak was at the bottom of the tube (in aggregates). Consistent with those findings, the LPL in the low-salt peak, but not that in the high-salt peak, was easily detectable with single mAb sandwich ELISAs, in which LPL is captured and detected with the same antibody. We conclude that catalytically active LPL can exist in a monomeric state.
Keywords: lipase; lipolysis; triglycerides.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: M. Meiyappan and J.E.G. are employees of Shire, now part of Takeda, and Takeda stock owners. K.M. is an employee of Immunobiologic Laboratories and holds stock in that company. K.N. holds stock in Immunobiologic Laboratories and serves as a consultant for Skylight and Sysmex.
Figures
Similar articles
-
Tamoxifen might influence the affinity of LPL for heparin-sepharose.Clin Chim Acta. 2003 Nov;337(1-2):141-5. doi: 10.1016/j.cccn.2003.08.006. Clin Chim Acta. 2003. PMID: 14568190
-
Lipoprotein lipase in human plasma is mainly inactive and associated with cholesterol-rich lipoproteins.J Lipid Res. 1993 Sep;34(9):1555-64. J Lipid Res. 1993. PMID: 8228638
-
Structure of lipoprotein lipase in complex with GPIHBP1.Proc Natl Acad Sci U S A. 2019 May 21;116(21):10360-10365. doi: 10.1073/pnas.1820171116. Epub 2019 May 9. Proc Natl Acad Sci U S A. 2019. PMID: 31072929 Free PMC article.
-
[Lipoprotein lipase].Nihon Rinsho. 1994 Dec;52(12):3146-52. Nihon Rinsho. 1994. PMID: 7853703 Review. Japanese.
-
Macromolecular Interactions of Lipoprotein Lipase (LPL).Subcell Biochem. 2024;104:139-179. doi: 10.1007/978-3-031-58843-3_8. Subcell Biochem. 2024. PMID: 38963487 Review.
Cited by
-
Using Synthetic ApoC-II Peptides and nAngptl4 Fragments to Measure Lipoprotein Lipase Activity in Radiometric and Fluorescent Assays.Front Cardiovasc Med. 2022 Jul 14;9:926631. doi: 10.3389/fcvm.2022.926631. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35911520 Free PMC article.
-
Combined action of albumin and heparin regulates lipoprotein lipase oligomerization, stability, and ligand interactions.PLoS One. 2023 Apr 12;18(4):e0283358. doi: 10.1371/journal.pone.0283358. eCollection 2023. PLoS One. 2023. PMID: 37043509 Free PMC article.
-
Polysorbates degrading enzymes in biotherapeutics - a current status and future perspectives.Front Bioeng Biotechnol. 2025 Jan 10;12:1490276. doi: 10.3389/fbioe.2024.1490276. eCollection 2024. Front Bioeng Biotechnol. 2025. PMID: 39867473 Free PMC article. Review.
-
The role of noncoding RNAs in cancer lipid metabolism.Front Oncol. 2022 Nov 14;12:1026257. doi: 10.3389/fonc.2022.1026257. eCollection 2022. Front Oncol. 2022. PMID: 36452489 Free PMC article. Review.
-
Is There a Nonlinear Relationship between Serum Uric Acid and Lipids in a Hypertensive Population with eGFR ≥30 ml/min/1.73 m2? Findings from the China Hypertension Registry Study.Int J Endocrinol. 2020 Sep 18;2020:9725979. doi: 10.1155/2020/9725979. eCollection 2020. Int J Endocrinol. 2020. PMID: 33014048 Free PMC article.
References
-
- Lookene A, Groot NB, Kastelein JJ, Olivecrona G, Bruin T. Mutation of tryptophan residues in lipoprotein lipase: Effects on stability, immunoreactivity, and catalytic properties. J Biol Chem. 1997;272:766–772. - PubMed
