

Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype
- PMID: 30850595
- PMCID: PMC6408494
- DOI: 10.1038/s41467-019-08989-2
Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype
Abstract
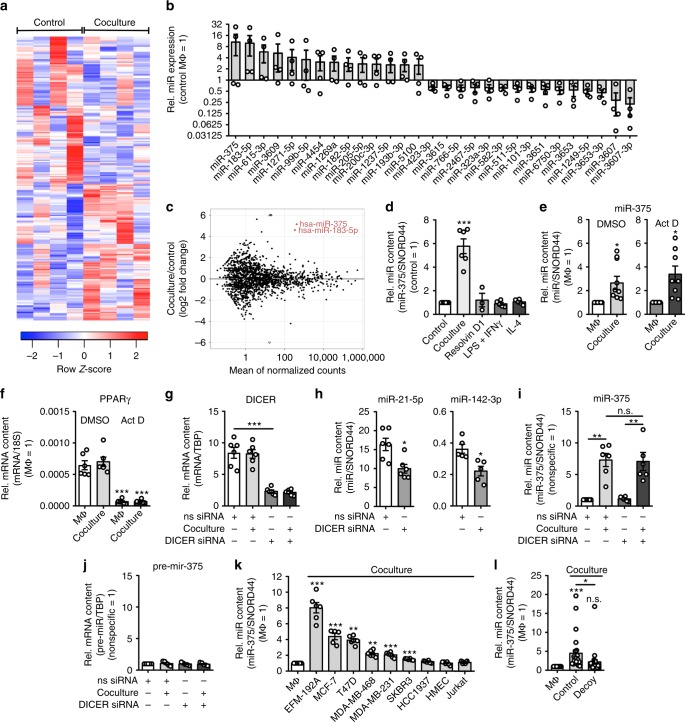
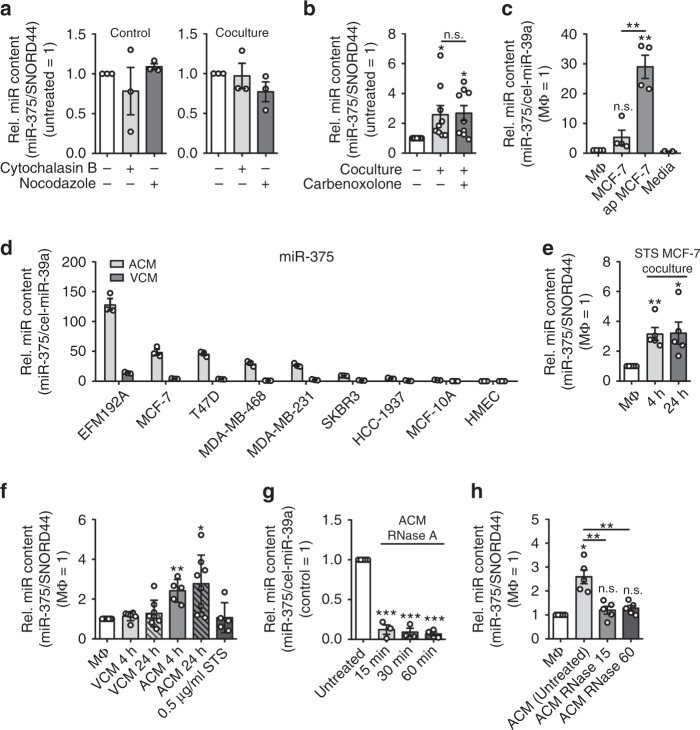
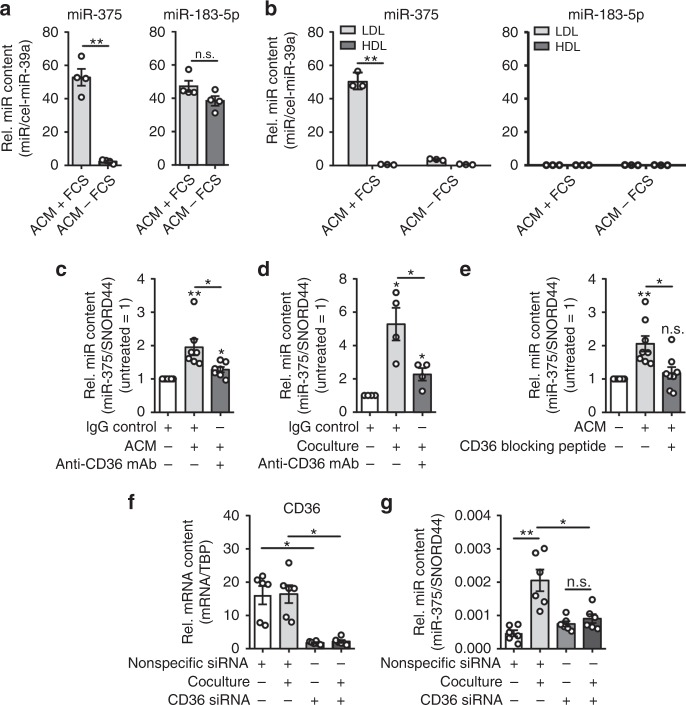
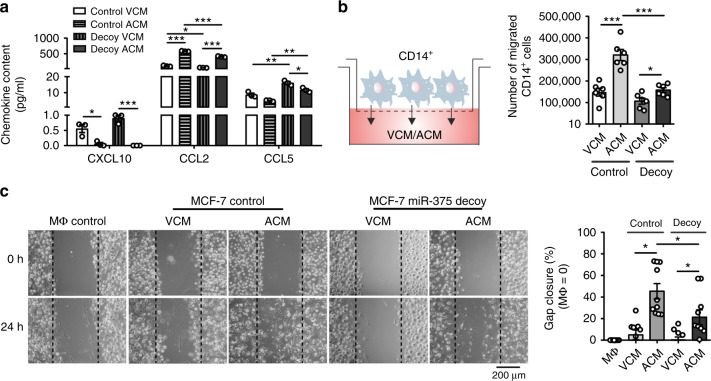
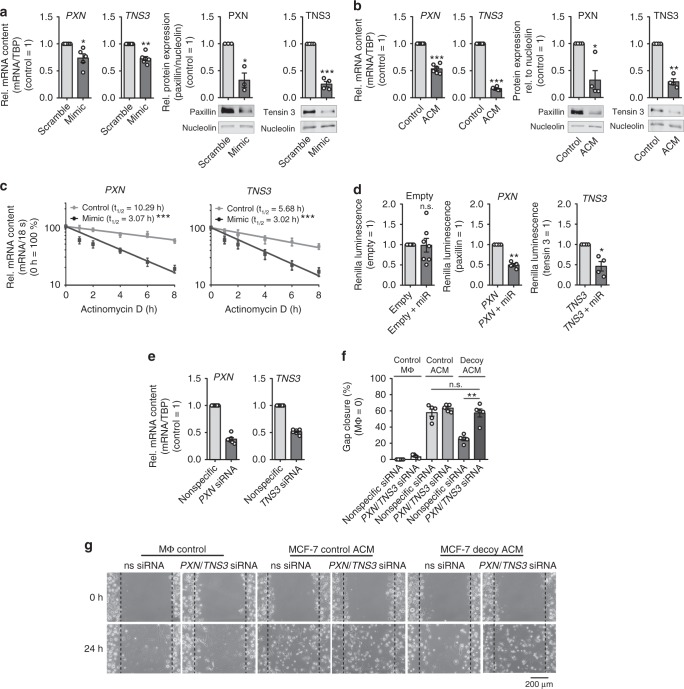
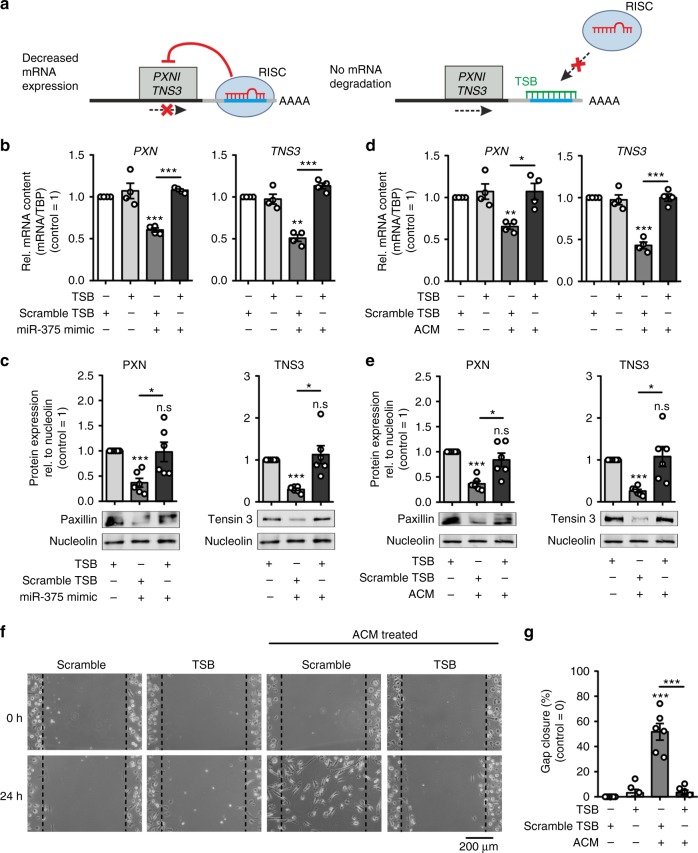
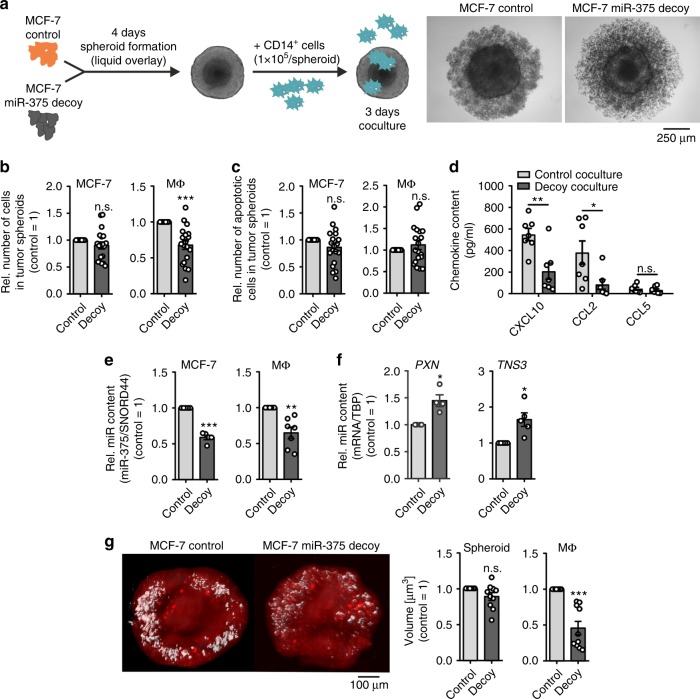
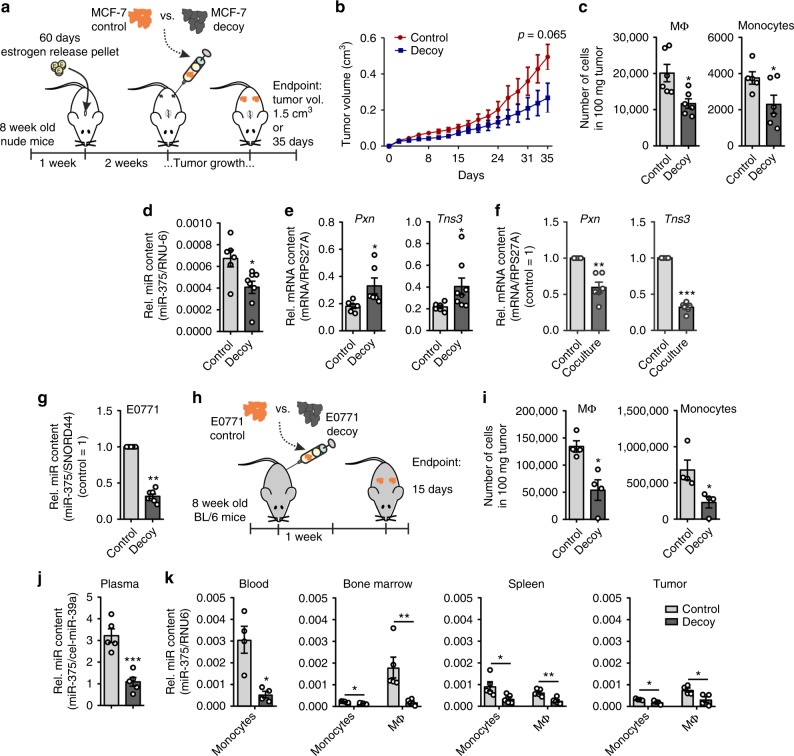
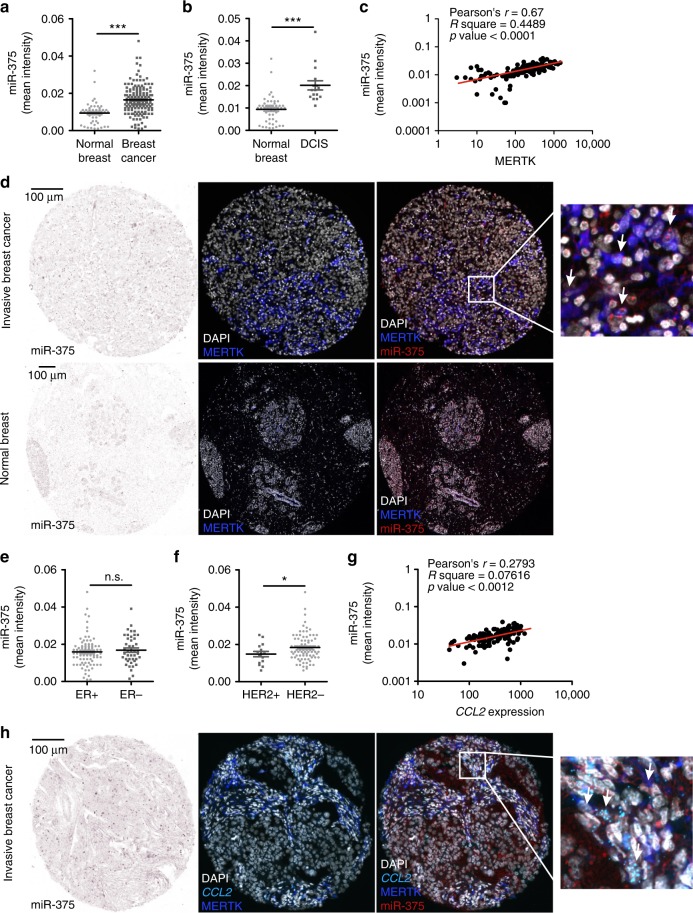
Tumor-immune cell interactions shape the immune cell phenotype, with microRNAs (miRs) being crucial components of this crosstalk. How they are transferred and how they affect their target landscape, especially in tumor-associated macrophages (TAMs), is largely unknown. Here we report that breast cancer cells have a high constitutive expression of miR-375, which is released as a non-exosome entity during apoptosis. Deep sequencing of the miRome pointed to enhanced accumulation of miR-375 in TAMs, facilitated by the uptake of tumor-derived miR-375 via CD36. In macrophages, miR-375 directly targets TNS3 and PXN to enhance macrophage migration and infiltration into tumor spheroids and in tumors of a xenograft mouse model. In tumor cells, miR-375 regulates CCL2 expression to increase recruitment of macrophages. Our study provides evidence for miR transfer from tumor cells to TAMs and identifies miR-375 as a crucial regulator of phagocyte infiltration and the subsequent development of a tumor-promoting microenvironment.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
MicroRNA-A Tumor Trojan Horse for Tumor-Associated Macrophages.Cells. 2019 Nov 21;8(12):1482. doi: 10.3390/cells8121482. Cells. 2019. PMID: 31766495 Free PMC article. Review.
-
Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis.Mol Cancer. 2019 Mar 30;18(1):64. doi: 10.1186/s12943-019-0976-4. Mol Cancer. 2019. PMID: 30927925 Free PMC article.
-
A double feedback loop mediated by microRNA-23a/27a/24-2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression.Oncotarget. 2016 Mar 22;7(12):13502-19. doi: 10.18632/oncotarget.6284. Oncotarget. 2016. PMID: 26540574 Free PMC article.
-
LincRNA-p21 knockdown reversed tumor-associated macrophages function by promoting MDM2 to antagonize* p53 activation and alleviate breast cancer development.Cancer Immunol Immunother. 2020 May;69(5):835-846. doi: 10.1007/s00262-020-02511-0. Epub 2020 Feb 15. Cancer Immunol Immunother. 2020. PMID: 32062693 Free PMC article.
-
MicroRNA-mediated control of macrophages and its implications for cancer.Trends Immunol. 2013 Jul;34(7):350-9. doi: 10.1016/j.it.2013.02.003. Epub 2013 Mar 13. Trends Immunol. 2013. PMID: 23498847 Free PMC article. Review.
Cited by
-
MicroRNAs: As Critical Regulators of Tumor- Associated Macrophages.Int J Mol Sci. 2020 Sep 27;21(19):7117. doi: 10.3390/ijms21197117. Int J Mol Sci. 2020. PMID: 32992449 Free PMC article. Review.
-
lncRNA DLX6-AS1 Promotes Myocardial Ischemia-Reperfusion Injury through Mediating the miR-204-5p/FBXW7 Axis.Mediators Inflamm. 2023 Jan 10;2023:9380398. doi: 10.1155/2023/9380398. eCollection 2023. Mediators Inflamm. 2023. PMID: 36660176 Free PMC article.
-
MicroRNA-A Tumor Trojan Horse for Tumor-Associated Macrophages.Cells. 2019 Nov 21;8(12):1482. doi: 10.3390/cells8121482. Cells. 2019. PMID: 31766495 Free PMC article. Review.
-
Decreased exosome-delivered miR-486-5p is responsible for the peritoneal metastasis of gastric cancer cells by promoting EMT progress.World J Surg Oncol. 2021 Oct 23;19(1):312. doi: 10.1186/s12957-021-02381-5. World J Surg Oncol. 2021. PMID: 34686196 Free PMC article.
-
GSK3 modulation in acute lung injury, myocarditis and polycystic kidney disease-related aneurysm.Biochim Biophys Acta Mol Cell Res. 2020 Nov;1867(11):118798. doi: 10.1016/j.bbamcr.2020.118798. Epub 2020 Jul 18. Biochim Biophys Acta Mol Cell Res. 2020. PMID: 32693109 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
