

Prospects for pharmacological targeting of pseudokinases
- PMID: 30850748
- PMCID: PMC7813639
- DOI: 10.1038/s41573-019-0018-3
Prospects for pharmacological targeting of pseudokinases
Abstract
Pseudokinases are members of the protein kinase superfamily but signal primarily through noncatalytic mechanisms. Many pseudokinases contribute to the pathologies of human diseases, yet they remain largely unexplored as drug targets owing to challenges associated with modulation of their biological functions. Our understanding of the structure and physiological roles of pseudokinases has improved substantially over the past decade, revealing intriguing similarities between pseudokinases and their catalytically active counterparts. Pseudokinases often adopt conformations that are analogous to those seen in catalytically active kinases and, in some cases, can also bind metal cations and/or nucleotides. Several clinically approved kinase inhibitors have been shown to influence the noncatalytic functions of active kinases, providing hope that similar properties in pseudokinases could be pharmacologically regulated. In this Review, we discuss known roles of pseudokinases in disease, their unique structural features and the progress that has been made towards developing pseudokinase-directed therapeutics.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures
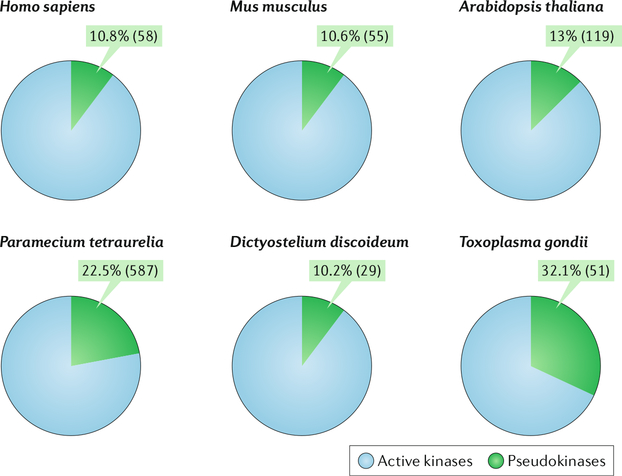
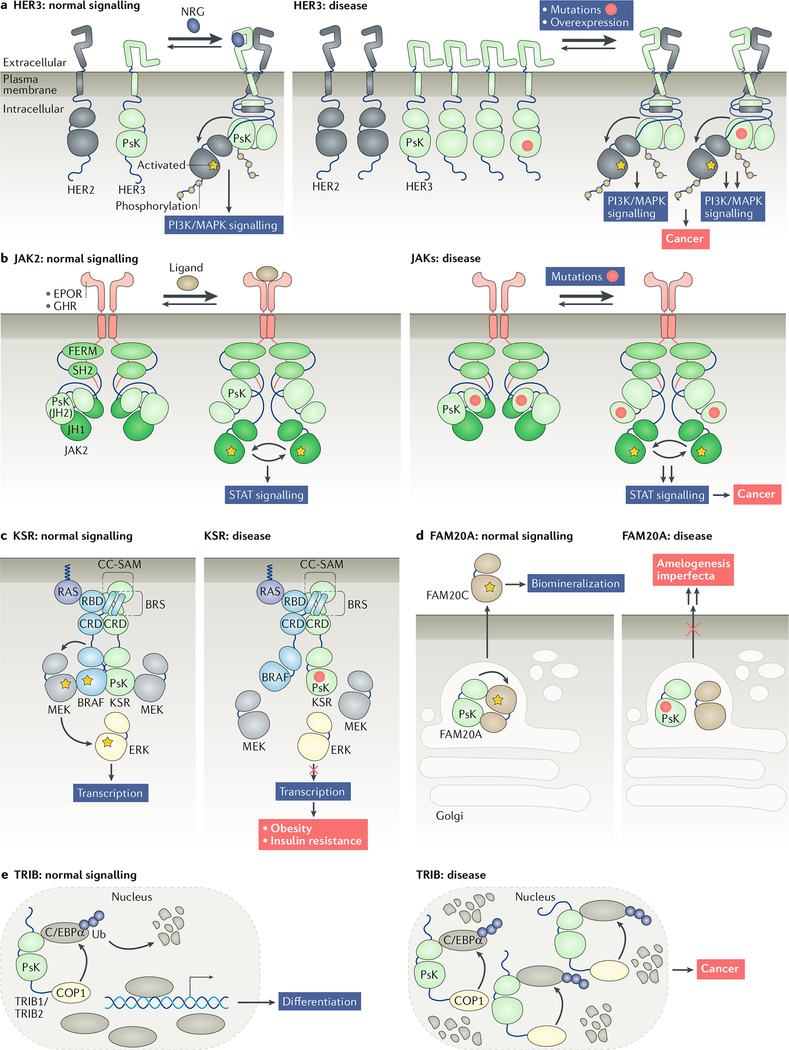
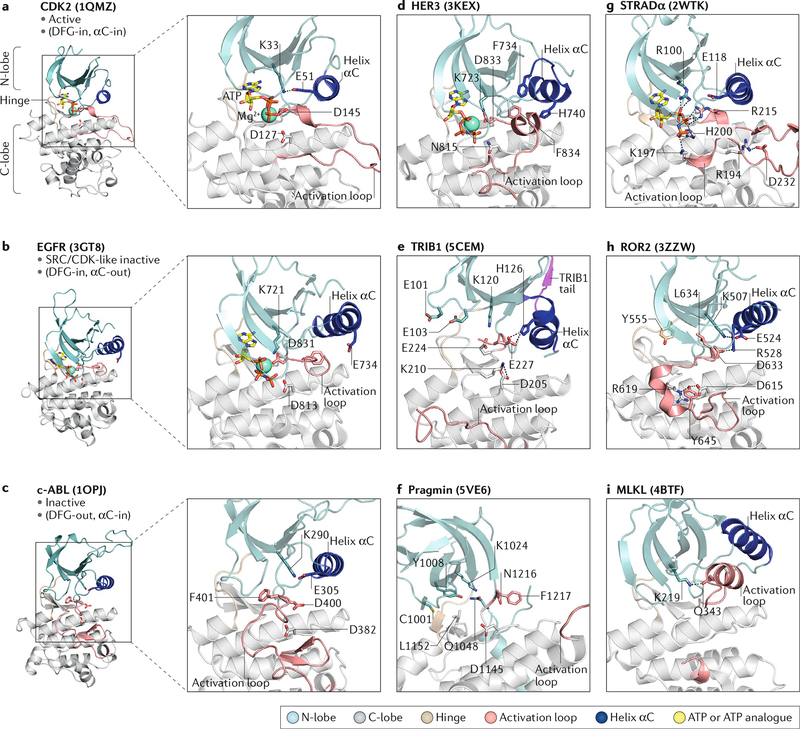

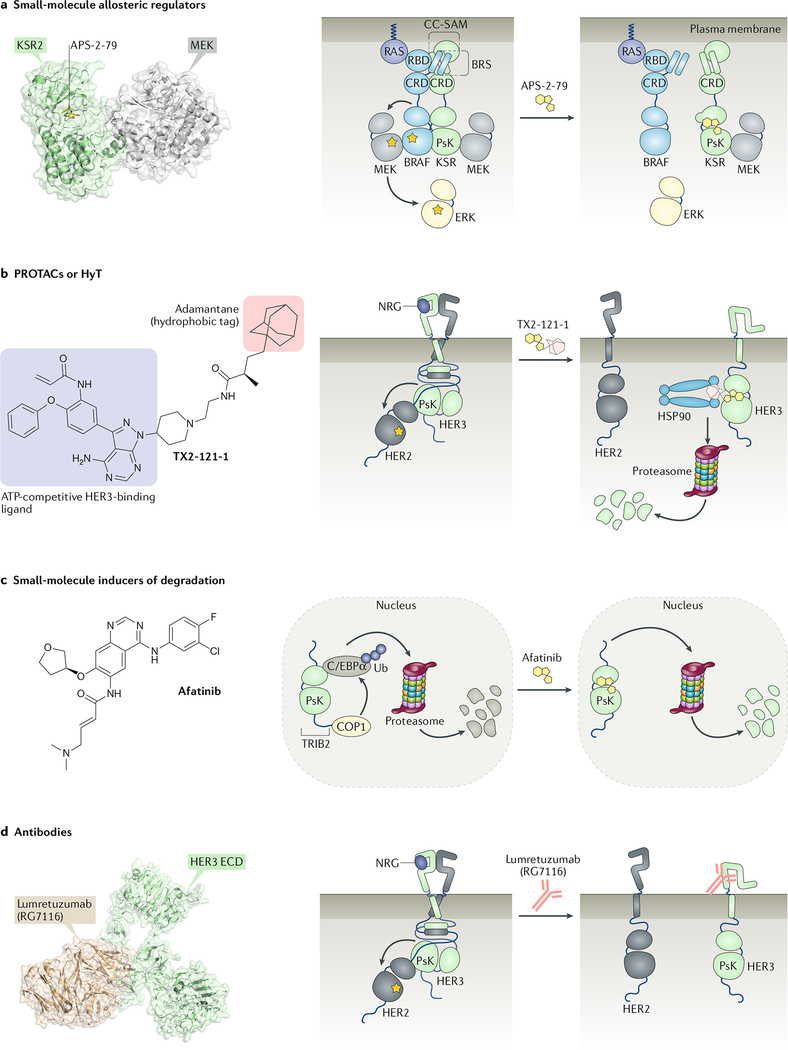
References
-
- Wu P, Nielsen TE & Clausen MH FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 36, 422–439 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
