

Interoperable and scalable data analysis with microservices: applications in metabolomics
- PMID: 30851093
- PMCID: PMC6761976
- DOI: 10.1093/bioinformatics/btz160
Interoperable and scalable data analysis with microservices: applications in metabolomics
Abstract
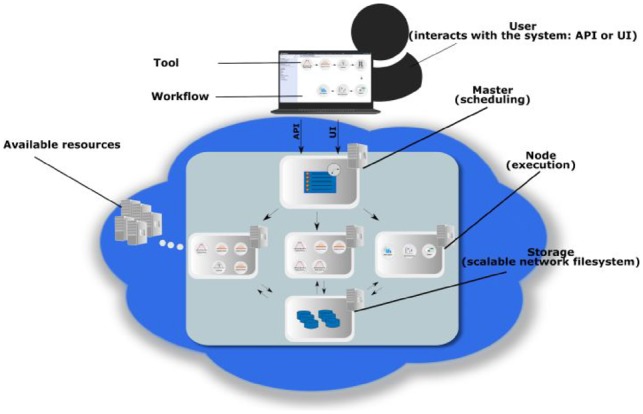
Motivation: Developing a robust and performant data analysis workflow that integrates all necessary components whilst still being able to scale over multiple compute nodes is a challenging task. We introduce a generic method based on the microservice architecture, where software tools are encapsulated as Docker containers that can be connected into scientific workflows and executed using the Kubernetes container orchestrator.
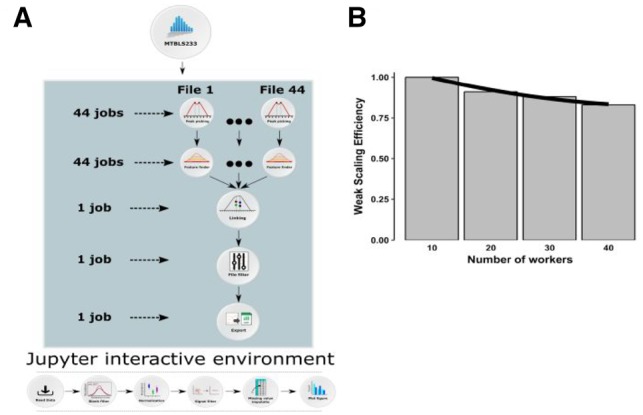
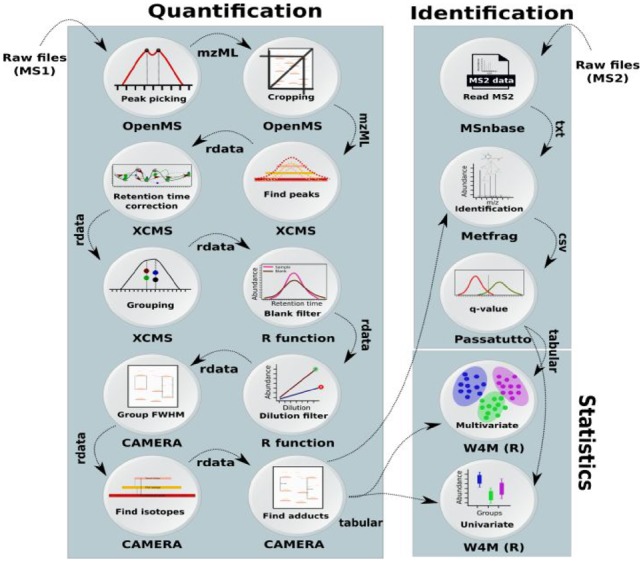
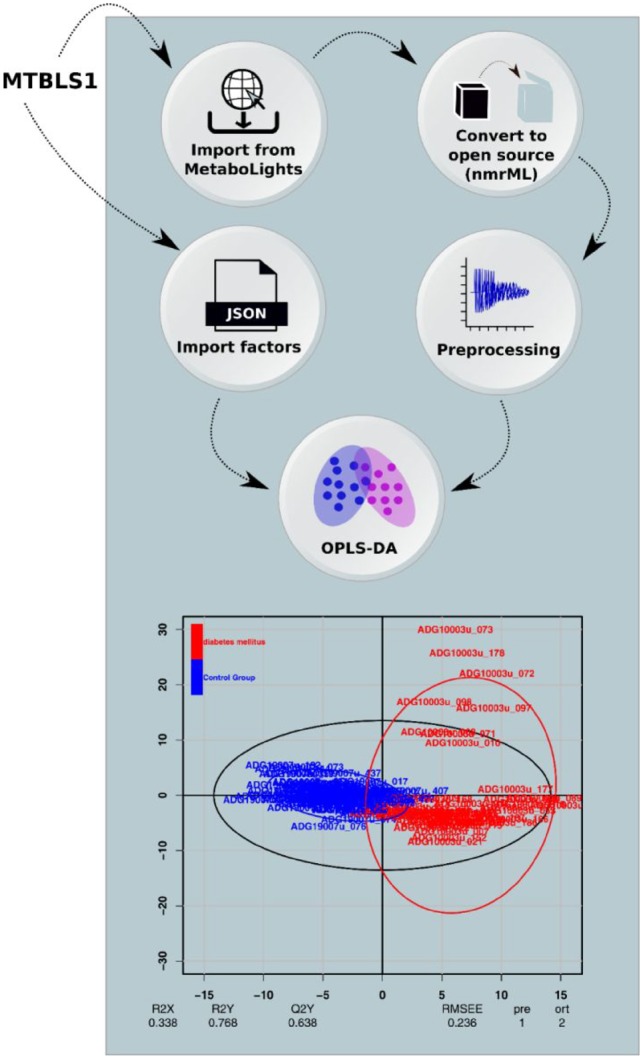
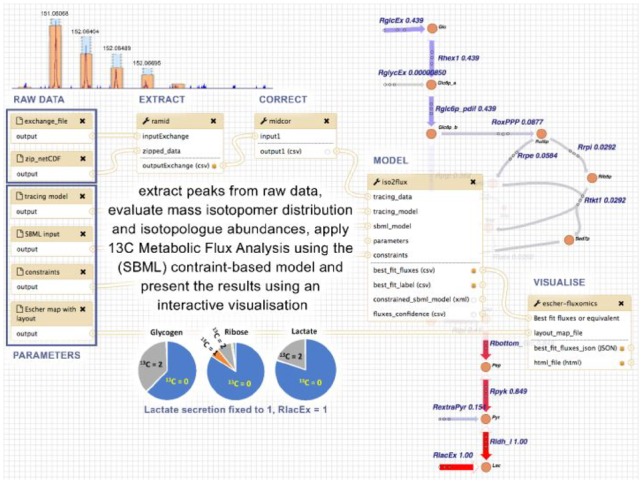
Results: We developed a Virtual Research Environment (VRE) which facilitates rapid integration of new tools and developing scalable and interoperable workflows for performing metabolomics data analysis. The environment can be launched on-demand on cloud resources and desktop computers. IT-expertise requirements on the user side are kept to a minimum, and workflows can be re-used effortlessly by any novice user. We validate our method in the field of metabolomics on two mass spectrometry, one nuclear magnetic resonance spectroscopy and one fluxomics study. We showed that the method scales dynamically with increasing availability of computational resources. We demonstrated that the method facilitates interoperability using integration of the major software suites resulting in a turn-key workflow encompassing all steps for mass-spectrometry-based metabolomics including preprocessing, statistics and identification. Microservices is a generic methodology that can serve any scientific discipline and opens up for new types of large-scale integrative science.
Availability and implementation: The PhenoMeNal consortium maintains a web portal (https://portal.phenomenal-h2020.eu) providing a GUI for launching the Virtual Research Environment. The GitHub repository https://github.com/phnmnl/ hosts the source code of all projects.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2019. Published by Oxford University Press.
Figures
Similar articles
-
PhenoMeNal: processing and analysis of metabolomics data in the cloud.Gigascience. 2019 Feb 1;8(2):giy149. doi: 10.1093/gigascience/giy149. Gigascience. 2019. PMID: 30535405 Free PMC article.
-
Container-based bioinformatics with Pachyderm.Bioinformatics. 2019 Mar 1;35(5):839-846. doi: 10.1093/bioinformatics/bty699. Bioinformatics. 2019. PMID: 30101309 Free PMC article.
-
Scalable Data Analysis in Proteomics and Metabolomics Using BioContainers and Workflows Engines.Proteomics. 2020 May;20(9):e1900147. doi: 10.1002/pmic.201900147. Epub 2019 Dec 18. Proteomics. 2020. PMID: 31657527 Free PMC article. Review.
-
Workflow4Metabolomics (W4M): A User-Friendly Metabolomics Platform for Analysis of Mass Spectrometry and Nuclear Magnetic Resonance Data.Curr Protoc. 2025 Feb;5(2):e70095. doi: 10.1002/cpz1.70095. Curr Protoc. 2025. PMID: 39951023
-
The metaRbolomics Toolbox in Bioconductor and beyond.Metabolites. 2019 Sep 23;9(10):200. doi: 10.3390/metabo9100200. Metabolites. 2019. PMID: 31548506 Free PMC article. Review.
Cited by
-
On-demand virtual research environments using microservices.PeerJ Comput Sci. 2019 Nov 11;5:e232. doi: 10.7717/peerj-cs.232. eCollection 2019. PeerJ Comput Sci. 2019. PMID: 33816885 Free PMC article.
-
Tackling the Challenges of 21st-Century Open Science and Beyond: A Data Science Lab Approach.Patterns (N Y). 2020 Sep 17;1(7):100103. doi: 10.1016/j.patter.2020.100103. eCollection 2020 Oct 9. Patterns (N Y). 2020. PMID: 33205137 Free PMC article.
-
From biomedical cloud platforms to microservices: next steps in FAIR data and analysis.Sci Data. 2022 Sep 8;9(1):553. doi: 10.1038/s41597-022-01619-5. Sci Data. 2022. PMID: 36075919 Free PMC article.
-
Experience in Developing an FHIR Medical Data Management Platform to Provide Clinical Decision Support.Int J Environ Res Public Health. 2019 Dec 20;17(1):73. doi: 10.3390/ijerph17010073. Int J Environ Res Public Health. 2019. PMID: 31861851 Free PMC article.
-
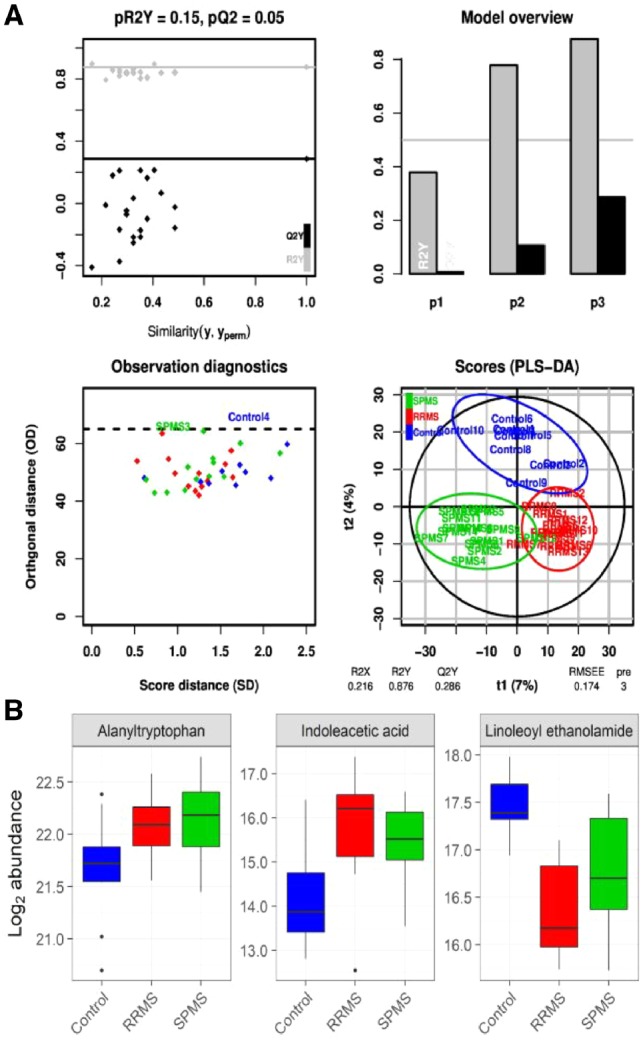
Integration of magnetic resonance imaging and protein and metabolite CSF measurements to enable early diagnosis of secondary progressive multiple sclerosis.Theranostics. 2018 Aug 7;8(16):4477-4490. doi: 10.7150/thno.26249. eCollection 2018. Theranostics. 2018. PMID: 30214633 Free PMC article.
References
-
- Allan R.N. (2009) Virtual Research Environments: From Portals to Science Gateways. ChandosŁ Publishing, Oxford, UK.
-
- Amirkhani A. et al. (2005) Interferon-beta affects the tryptophan metabolism in multiple sclerosis patients. Eur. J. Neurol., 12, 625–631. - PubMed
-
- Atkinson M. et al. (2017) Scientific workflows: past, present and future. Future Gener. Comput. Syst., 75, 216–227.
-
- Baker D., Pryce G. (2008) The endocannabinoid system and multiple sclerosis. Curr. Pharm. Des., 14, 2326–2336. - PubMed
Publication types
MeSH terms
Grants and funding
- 001/WHO_/World Health Organization/International
- BB/H024921/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I000771/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/L024055/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
