

Bayesian Detection of Convergent Rate Changes of Conserved Noncoding Elements on Phylogenetic Trees
- PMID: 30851112
- PMCID: PMC6501877
- DOI: 10.1093/molbev/msz049
Bayesian Detection of Convergent Rate Changes of Conserved Noncoding Elements on Phylogenetic Trees
Abstract
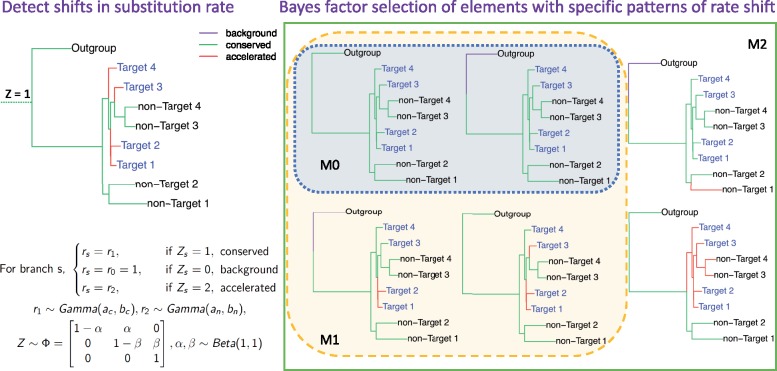
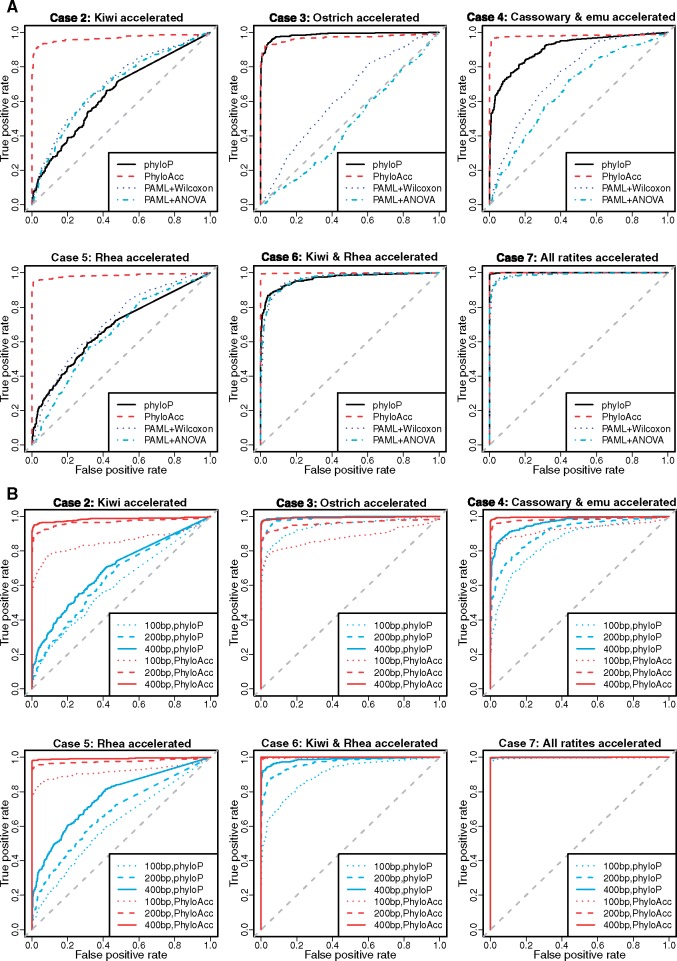
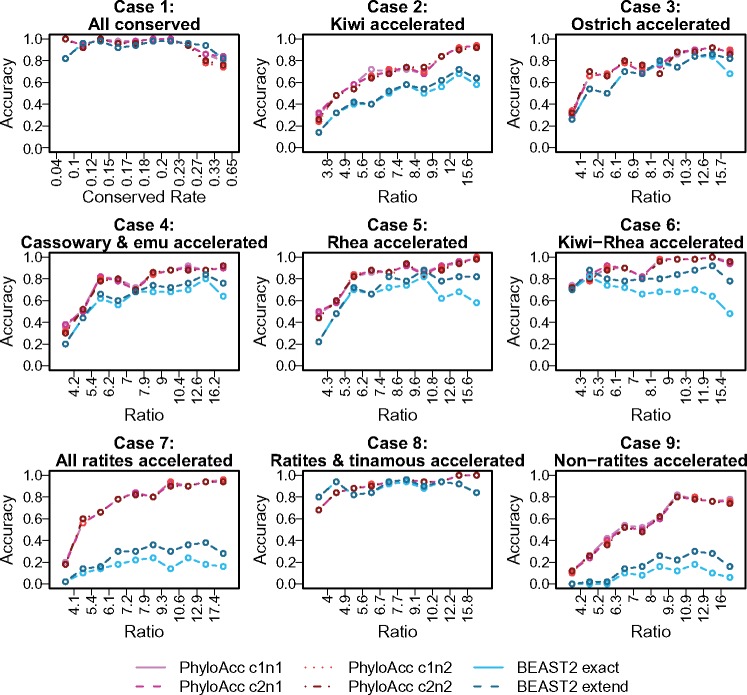
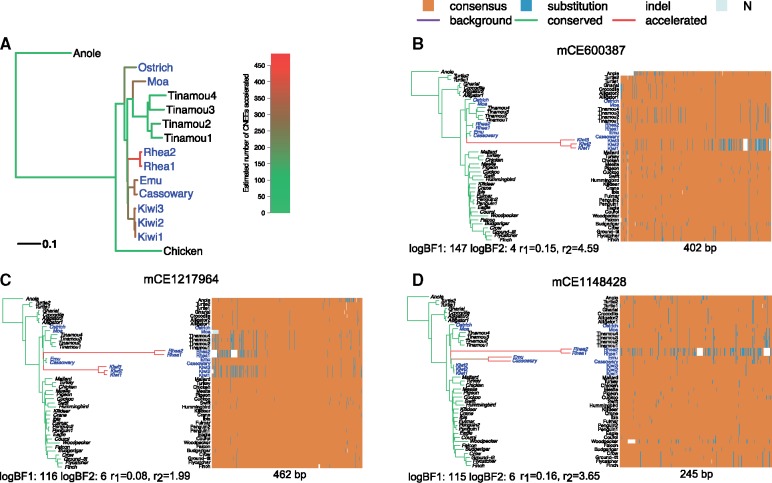
Conservation of DNA sequence over evolutionary time is a strong indicator of function, and gain or loss of sequence conservation can be used to infer changes in function across a phylogeny. Changes in evolutionary rates on particular lineages in a phylogeny can indicate shared functional shifts, and thus can be used to detect genomic correlates of phenotypic convergence. However, existing methods do not allow easy detection of patterns of rate variation, which causes challenges for detecting convergent rate shifts or other complex evolutionary scenarios. Here we introduce PhyloAcc, a new Bayesian method to model substitution rate changes in conserved elements across a phylogeny. The method assumes several categories of substitution rate for each branch on the phylogenetic tree, estimates substitution rates per category, and detects changes of substitution rate as the posterior probability of a category switch. Simulations show that PhyloAcc can detect genomic regions with rate shifts in multiple target species better than previous methods and has a higher accuracy of reconstructing complex patterns of substitution rate changes than prevalent Bayesian relaxed clock models. We demonstrate the utility of PhyloAcc in two classic examples of convergent phenotypes: loss of flight in birds and the transition to marine life in mammals. In each case, our approach reveals numerous examples of conserved nonexonic elements with accelerations specific to the phenotypically convergent lineages. Our method is widely applicable to any set of conserved elements where multiple rate changes are expected on a phylogeny.
Keywords: Bayesian model; comparative genomics; convergence; mammal; phylogenetics.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Angelis K, dos Reis M.. 2015. The impact of ancestral population size and incomplete lineage sorting on Bayesian estimation of species divergence times. Curr Zool. 61(5): 874–885.
-
- Baker AJ, Haddrath O, McPherson JD, Cloutier A.. 2014. Genomic support for a moa-tinamou clade and adaptive morphological convergence in flightless ratites. Mol Biol Evol. 31(7): 1686–1696. - PubMed
-
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D.. 2004. Ultraconserved elements in the human genome. Science 304(5675): 1321–1325. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
