

Phylogenetic Clustering by Linear Integer Programming (PhyCLIP)
- PMID: 30854550
- PMCID: PMC6573476
- DOI: 10.1093/molbev/msz053
Phylogenetic Clustering by Linear Integer Programming (PhyCLIP)
Abstract
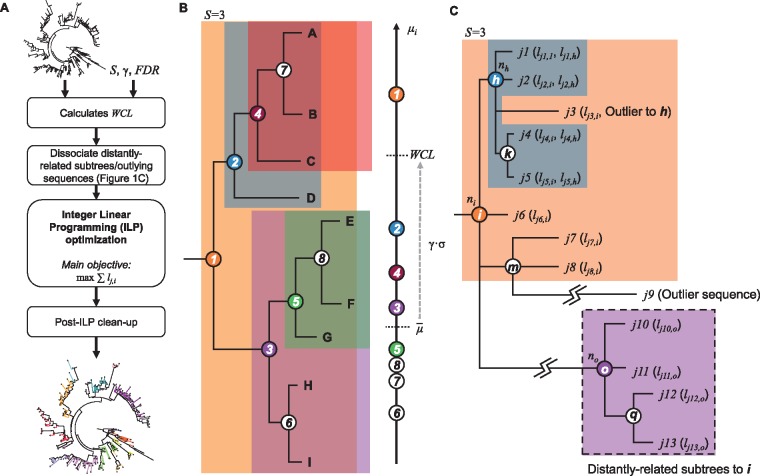
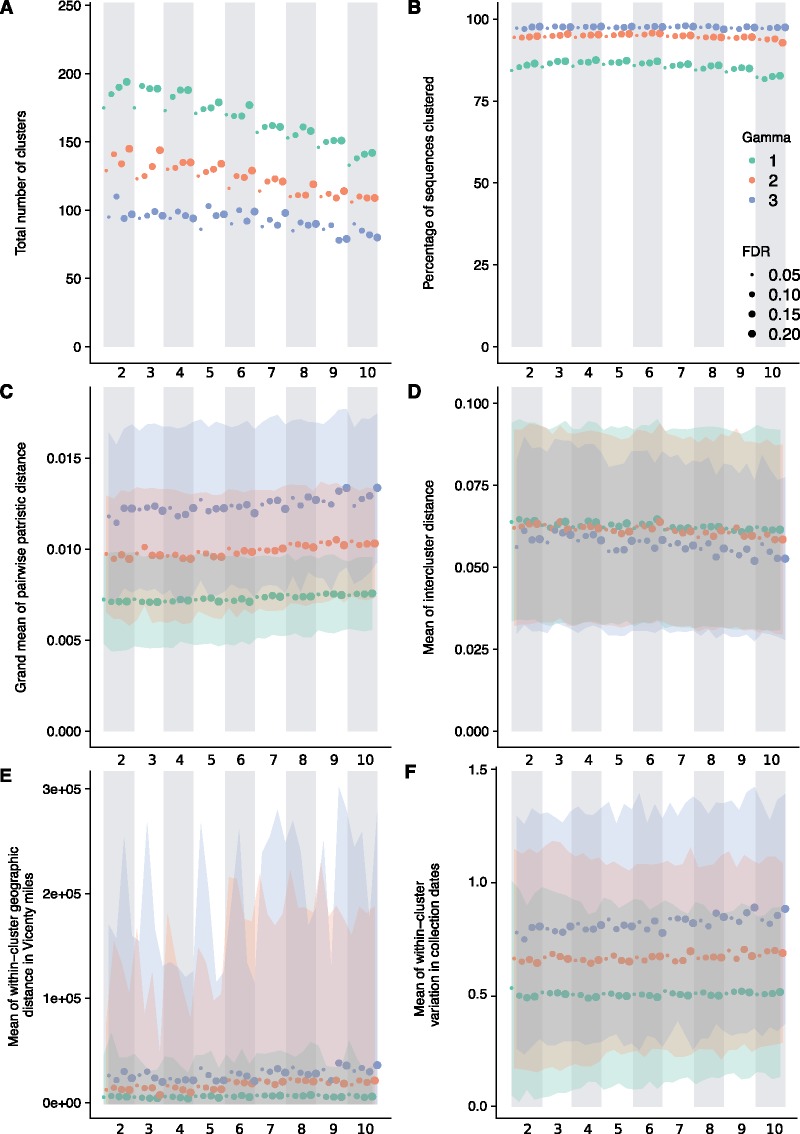
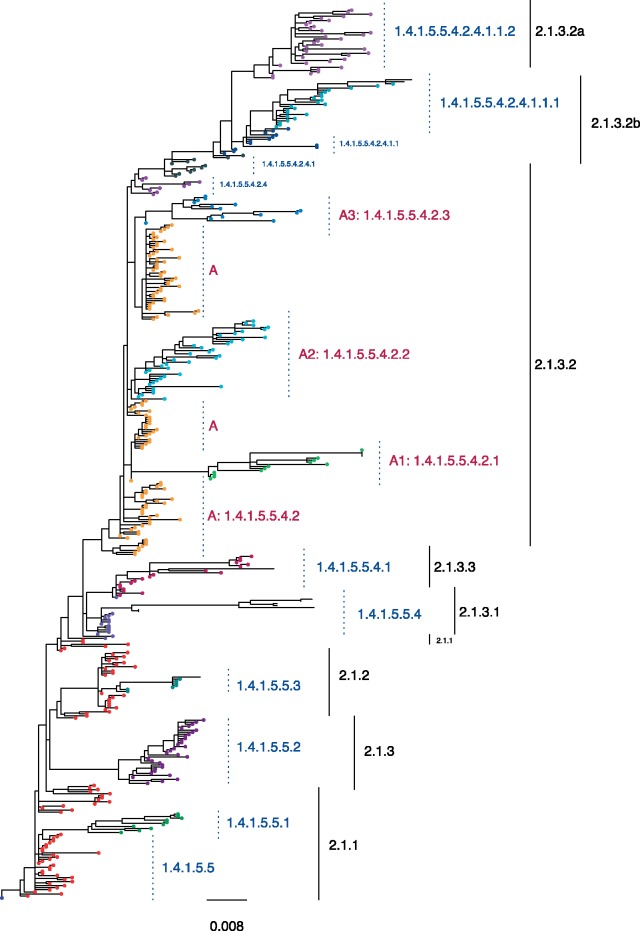
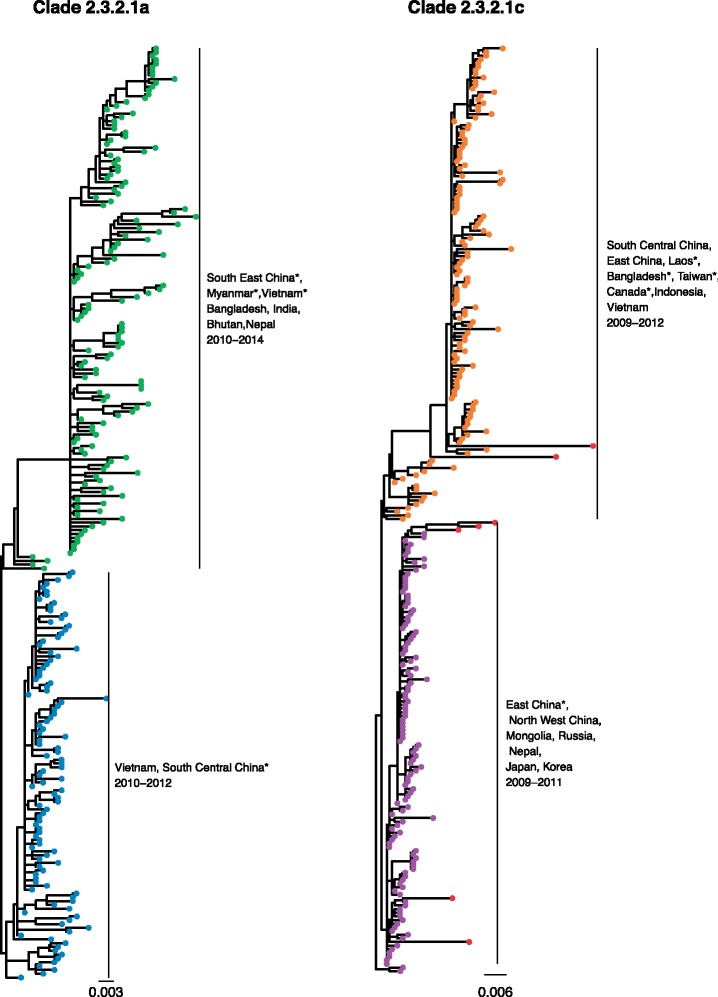
Subspecies nomenclature systems of pathogens are increasingly based on sequence data. The use of phylogenetics to identify and differentiate between clusters of genetically similar pathogens is particularly prevalent in virology from the nomenclature of human papillomaviruses to highly pathogenic avian influenza (HPAI) H5Nx viruses. These nomenclature systems rely on absolute genetic distance thresholds to define the maximum genetic divergence tolerated between viruses designated as closely related. However, the phylogenetic clustering methods used in these nomenclature systems are limited by the arbitrariness of setting intra and intercluster diversity thresholds. The lack of a consensus ground truth to define well-delineated, meaningful phylogenetic subpopulations amplifies the difficulties in identifying an informative distance threshold. Consequently, phylogenetic clustering often becomes an exploratory, ad hoc exercise. Phylogenetic Clustering by Linear Integer Programming (PhyCLIP) was developed to provide a statistically principled phylogenetic clustering framework that negates the need for an arbitrarily defined distance threshold. Using the pairwise patristic distance distributions of an input phylogeny, PhyCLIP parameterizes the intra and intercluster divergence limits as statistical bounds in an integer linear programming model which is subsequently optimized to cluster as many sequences as possible. When applied to the hemagglutinin phylogeny of HPAI H5Nx viruses, PhyCLIP was not only able to recapitulate the current WHO/OIE/FAO H5 nomenclature system but also further delineated informative higher resolution clusters that capture geographically distinct subpopulations of viruses. PhyCLIP is pathogen-agnostic and can be generalized to a wide variety of research questions concerning the identification of biologically informative clusters in pathogen phylogenies. PhyCLIP is freely available at http://github.com/alvinxhan/PhyCLIP, last accessed March 15, 2019.
Keywords: influenza; molecular epidemiology; nomenclature; pathogen; phylogenetic clustering.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Burk RD, Chen Z, Harari A, Smith BC, Kocjan BJ, Maver PJ, Poljak M.. 2011. Classification and nomenclature system for human Alphapapillomavirus variants: general features, nucleotide landmarks and assignment of HPV6 and HPV11 isolates to variant lineages. Acta Dermatovenerol Alp Pannonica Adriat. 20:113–123. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
