

Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling
- PMID: 30857233
- PMCID: PMC6468910
- DOI: 10.3390/cancers11030338
Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling
Abstract
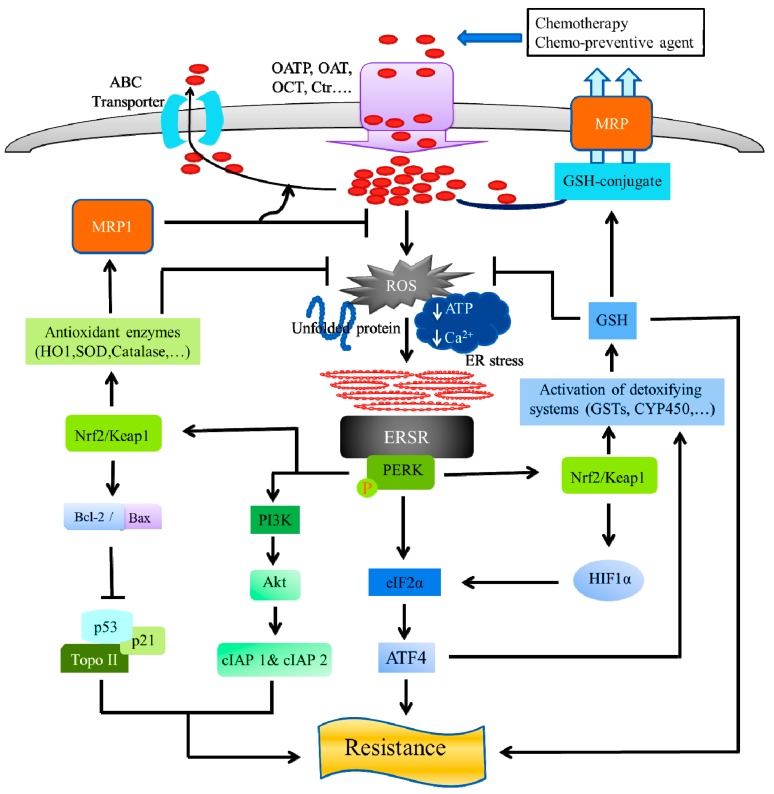
Cancers cells have the ability to develop chemotherapy resistance, which is a persistent problem during cancer treatment. Chemotherapy resistance develops through different molecular mechanisms, which lead to modification of the cancer cells signals needed for cellular proliferation or for stimulating an immune response. The endoplasmic reticulum (ER) is an important organelle involved in protein quality control, by promoting the correct folding of protein and ER-mediated degradation of unfolded or misfolded protein, namely, ER-associated degradation. Disturbances of the normal ER functions causes an accumulation of unfolded or misfolded proteins in the ER lumen, resulting in a condition called "ER stress (ERS)." ERS triggers the unfolded protein response (UPR)-also called the ERS response (ERSR)-to restore homeostasis or activate cell death. Although the ERSR is one emerging potential target for chemotherapeutics to treat cancer, it is also critical for chemotherapeutics resistance, as well. However, the detailed molecular mechanism of the relationship between the ERSR and tumor survival or drug resistance remains to be fully understood. In this review, we aim to describe the most vital molecular mechanism of the relationship between the ERSR and chemotherapy resistance. Moreover, the review also discusses the molecular mechanism of ER stress-mediated apoptosis on cancer treatments.
Keywords: cancer; chemotherapy resistance; endoplasmic reticulum; endoplasmic reticulum stress response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Heron M., Anderson R.N. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality. NCHS; Hyattsville, MD, USA: 2016. pp. 1–8. NCHS Data Brief 2016. - PubMed
-
- Korean Statistical Information Service National Cancer Registration Data. [(accessed on 6 July 2018)]; Available online: www.kosis.kr.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
