

Learning protein constitutive motifs from sequence data
- PMID: 30857591
- PMCID: PMC6436896
- DOI: 10.7554/eLife.39397
Learning protein constitutive motifs from sequence data
Abstract
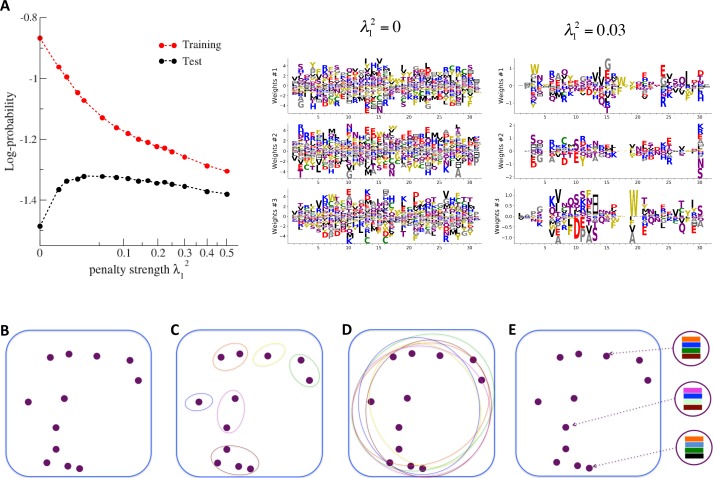
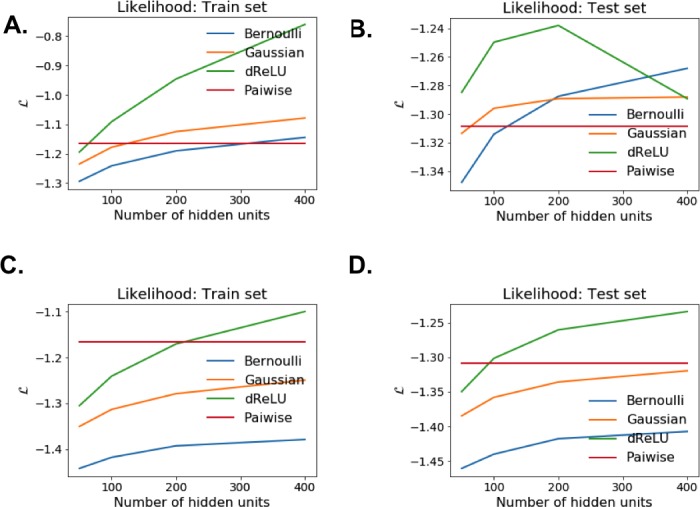
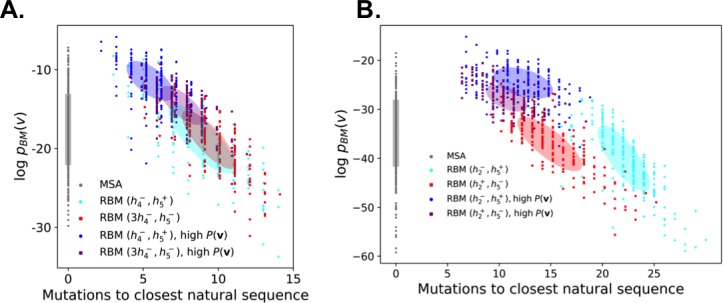
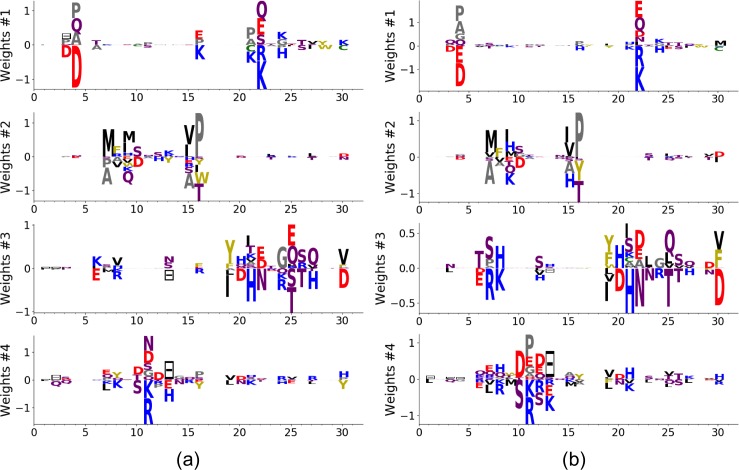
Statistical analysis of evolutionary-related protein sequences provides information about their structure, function, and history. We show that Restricted Boltzmann Machines (RBM), designed to learn complex high-dimensional data and their statistical features, can efficiently model protein families from sequence information. We here apply RBM to 20 protein families, and present detailed results for two short protein domains (Kunitz and WW), one long chaperone protein (Hsp70), and synthetic lattice proteins for benchmarking. The features inferred by the RBM are biologically interpretable: they are related to structure (residue-residue tertiary contacts, extended secondary motifs (α-helixes and β-sheets) and intrinsically disordered regions), to function (activity and ligand specificity), or to phylogenetic identity. In addition, we use RBM to design new protein sequences with putative properties by composing and 'turning up' or 'turning down' the different modes at will. Our work therefore shows that RBM are versatile and practical tools that can be used to unveil and exploit the genotype-phenotype relationship for protein families.
Keywords: coevolution; computational biology; machine learning; none; physics of living systems; sequence analysis; systems biology.
© 2019, Tubiana et al.
Conflict of interest statement
JT, SC, RM No competing interests declared
Figures























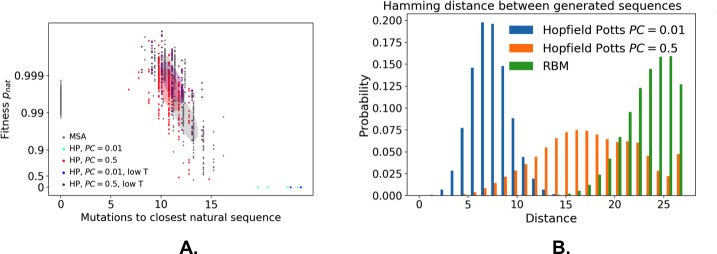








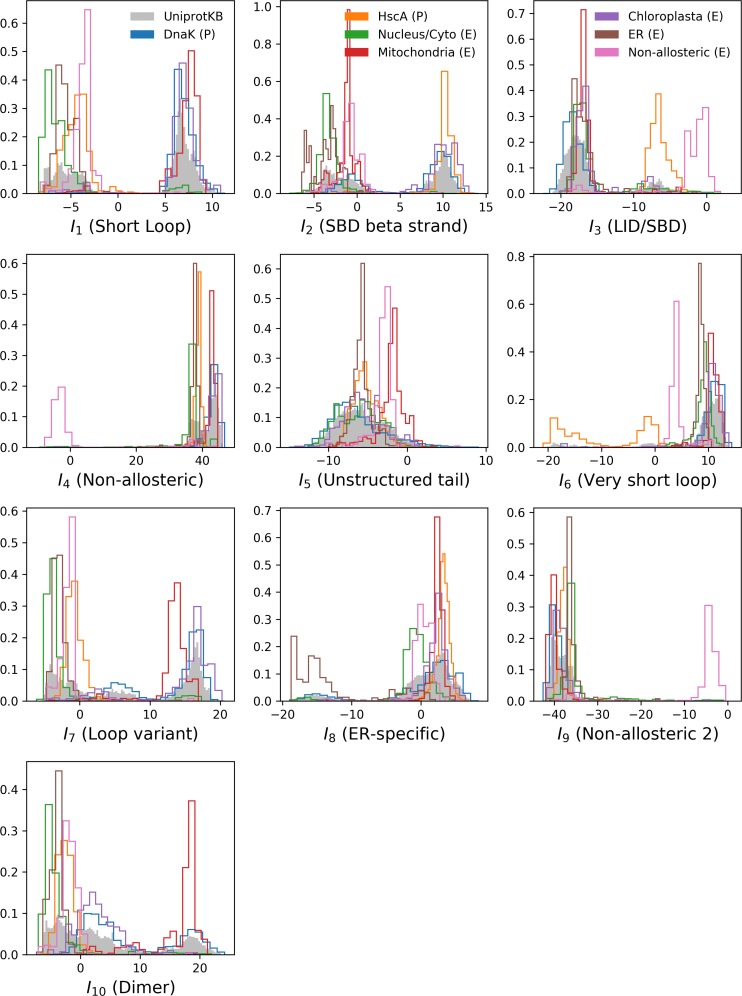


References
-
- Ackley DH, Hinton GE, Sejnowski TJ. Readings in Computer Vision. Elsevier; 1987. A learning algorithm for boltzmann machines; pp. 522–533.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
