

PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes
- PMID: 30858161
- PMCID: PMC6412922
- DOI: 10.26508/lsa.201900353
PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes
Abstract
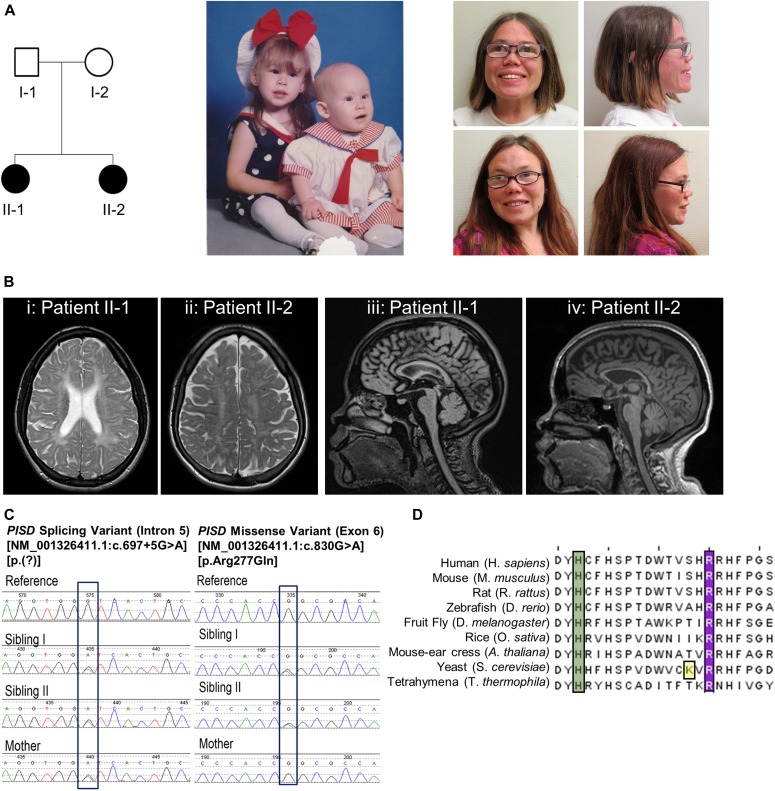
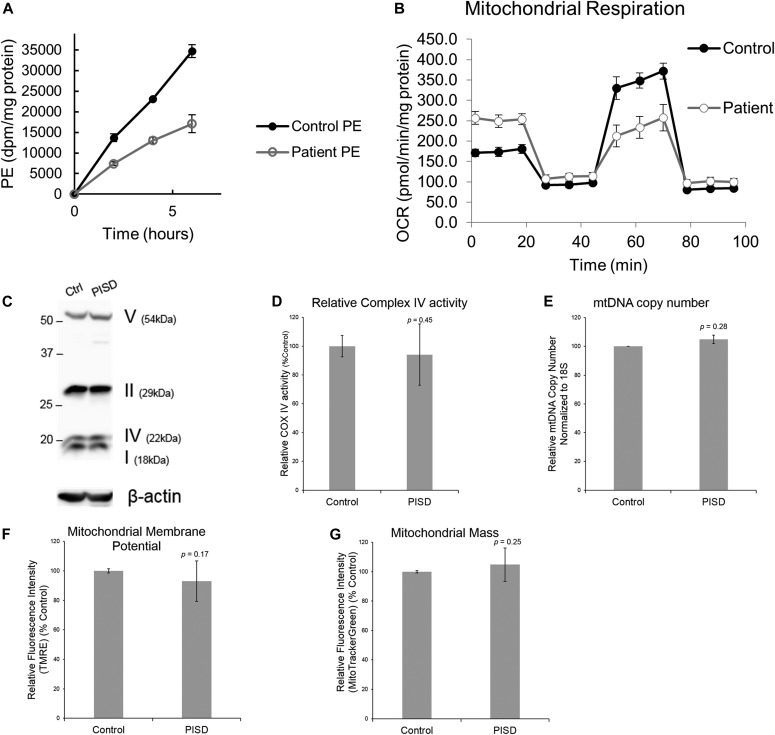
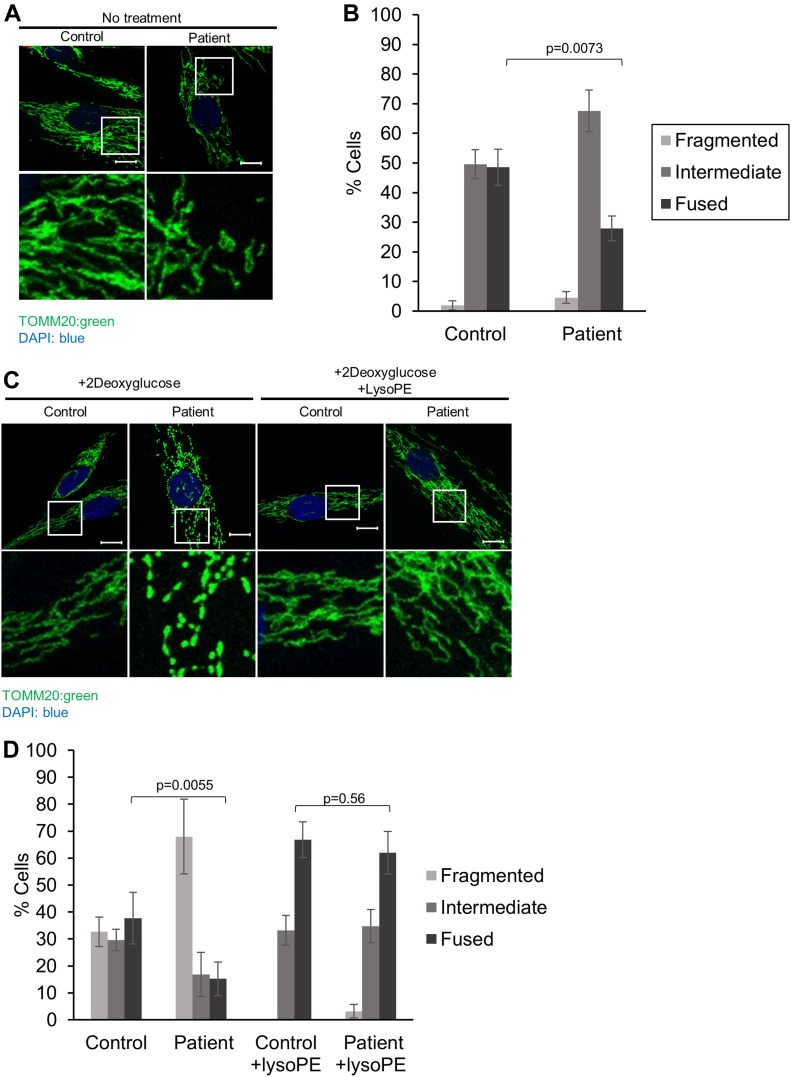
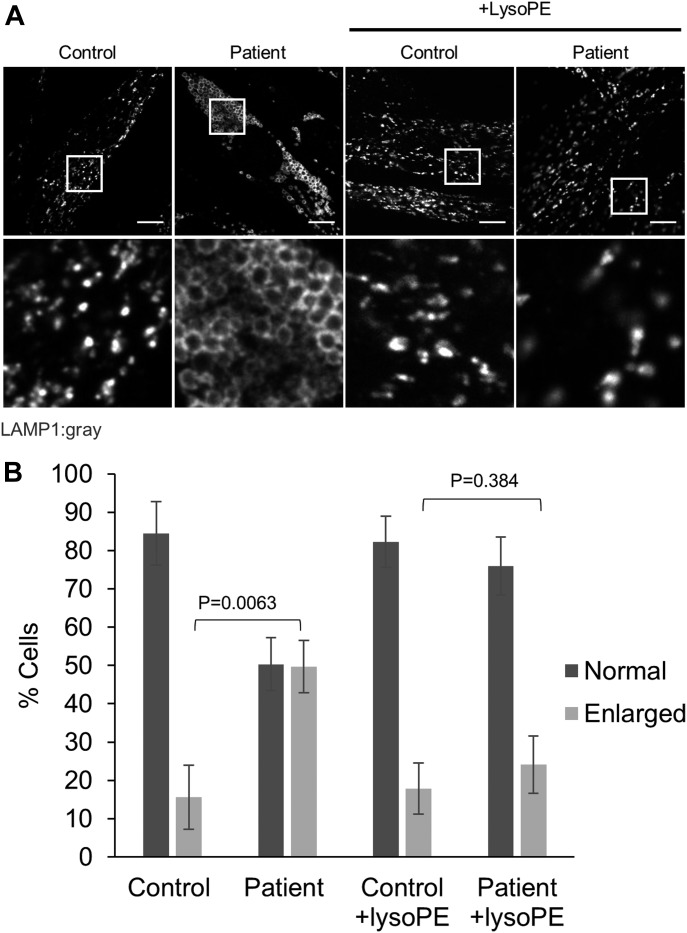
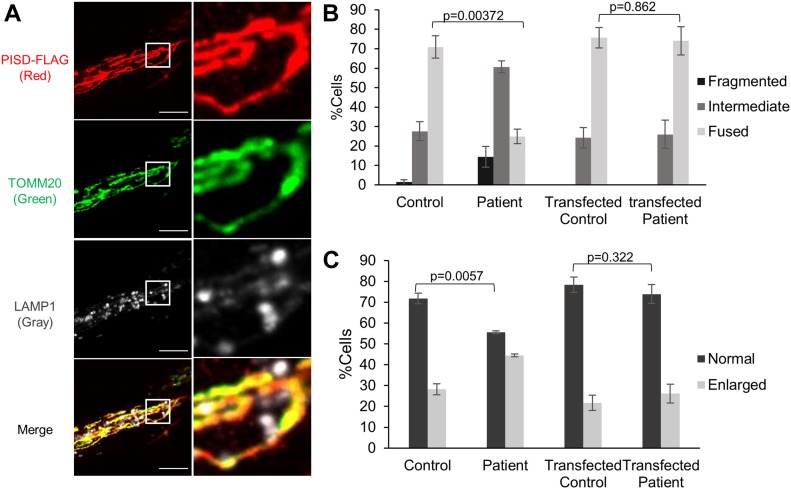
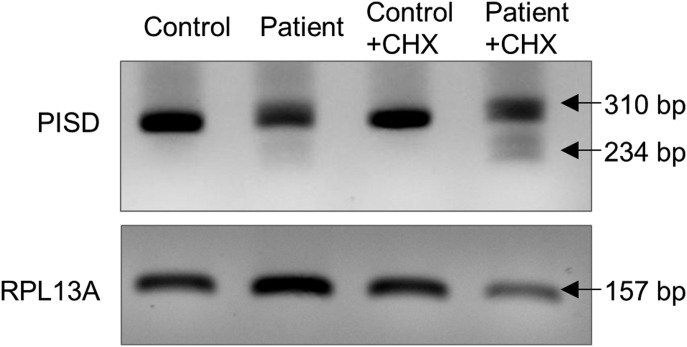
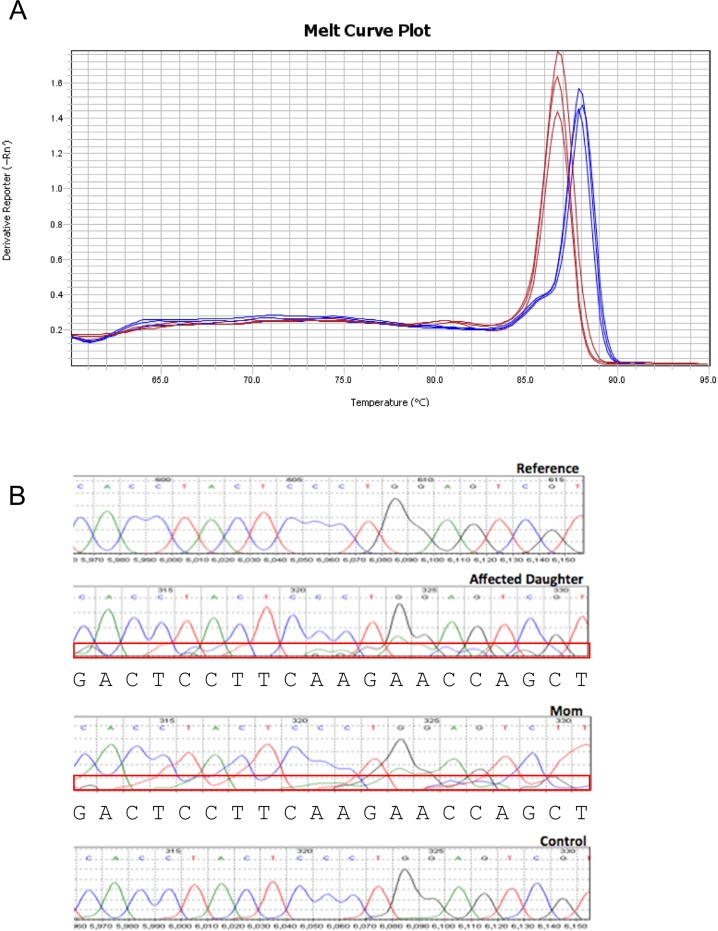
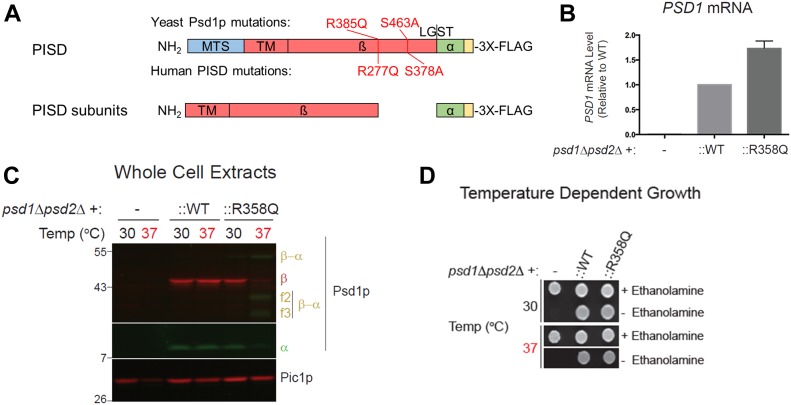
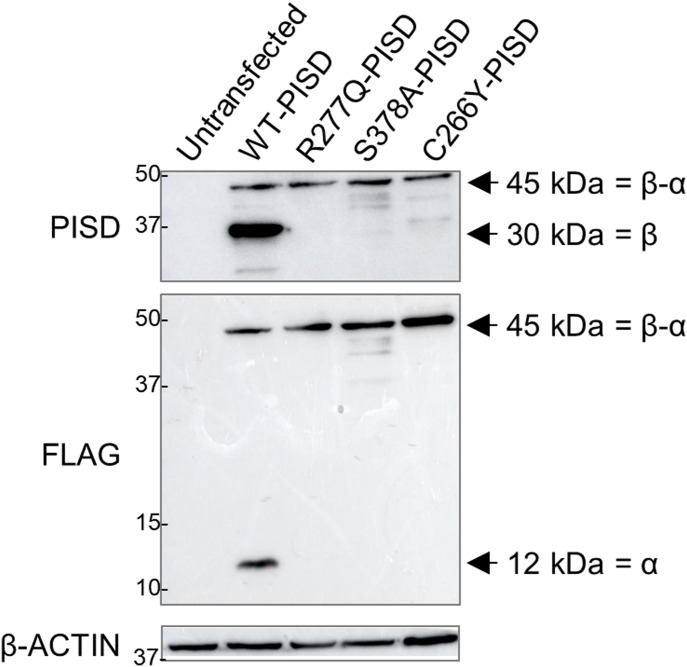
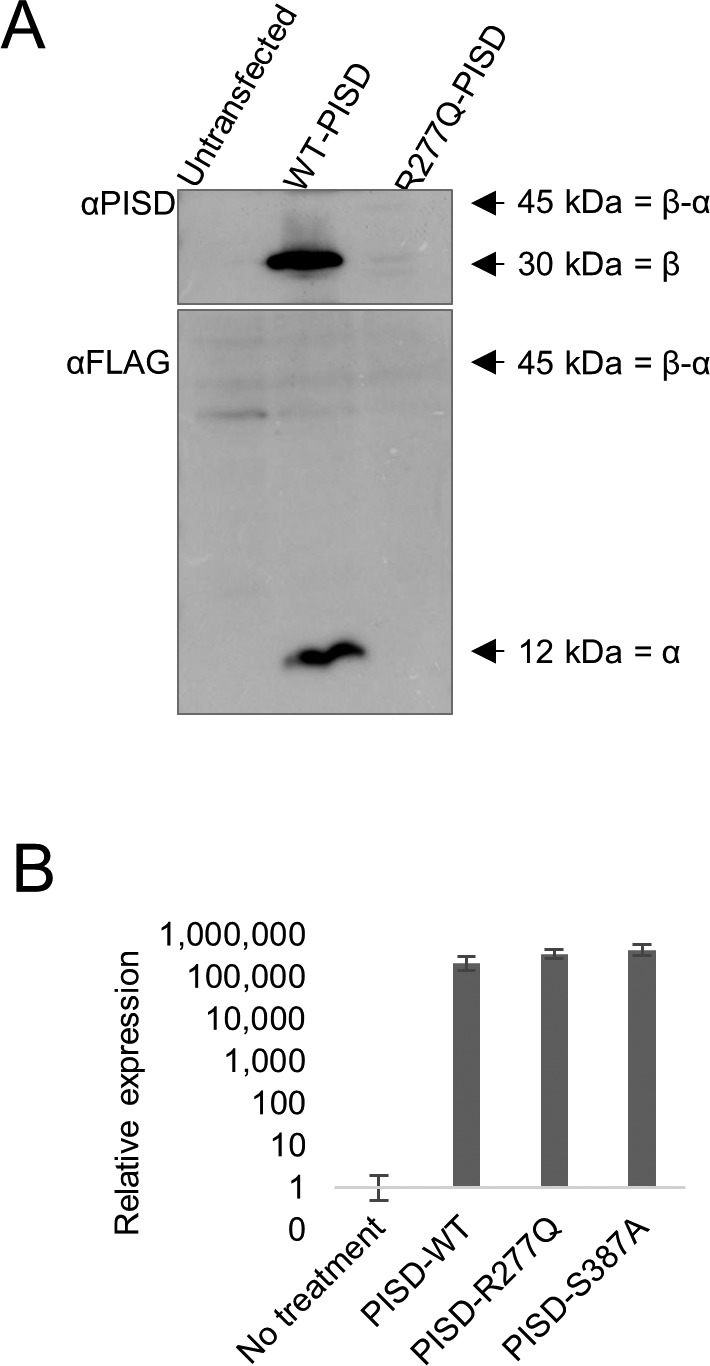
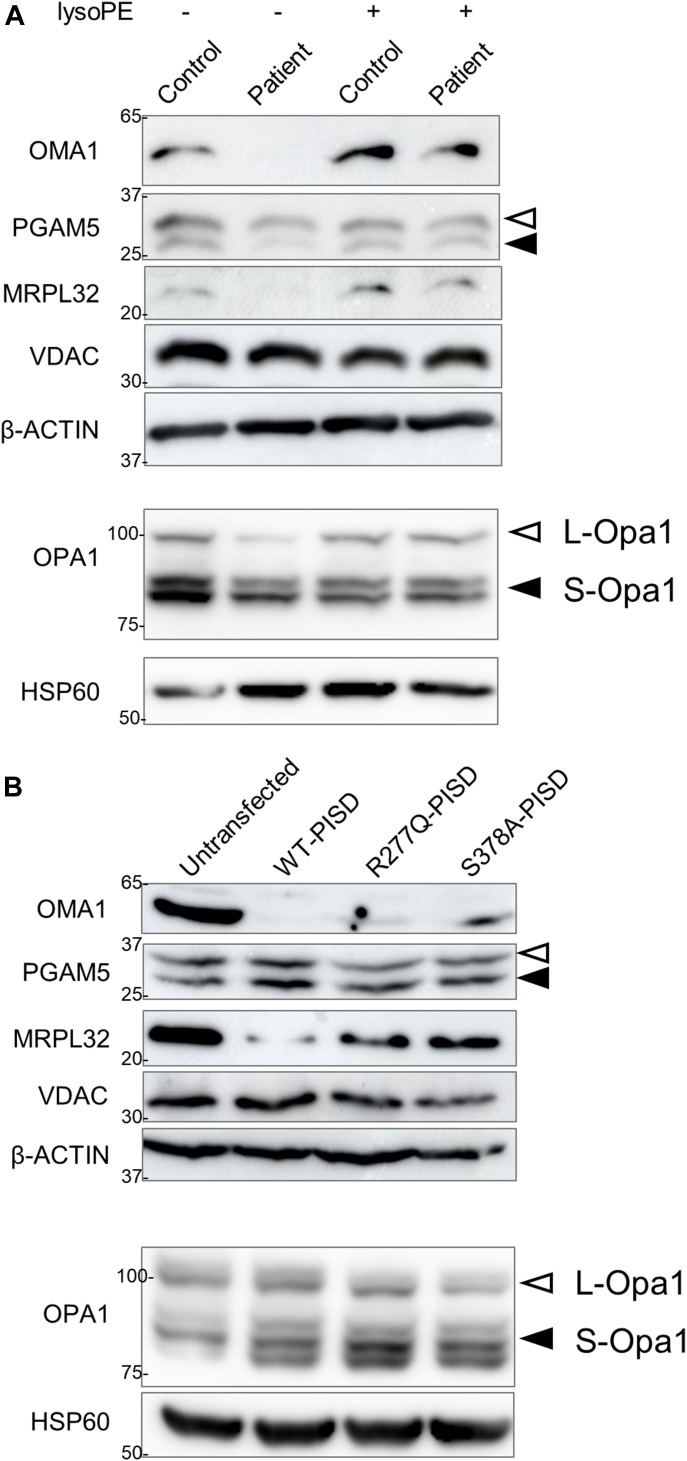
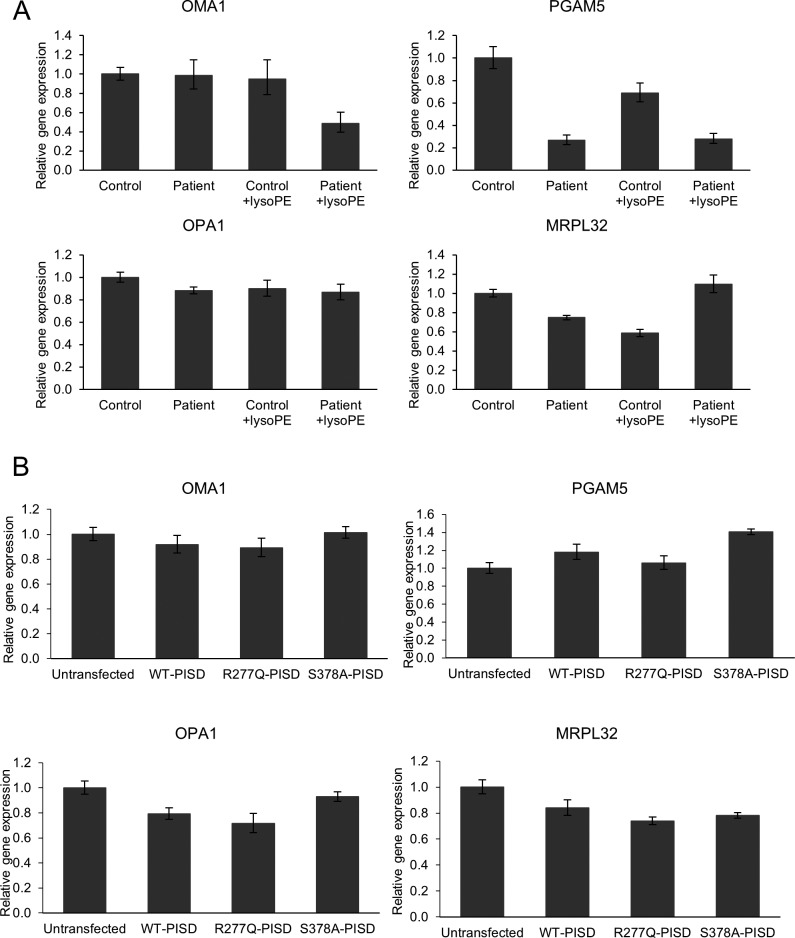
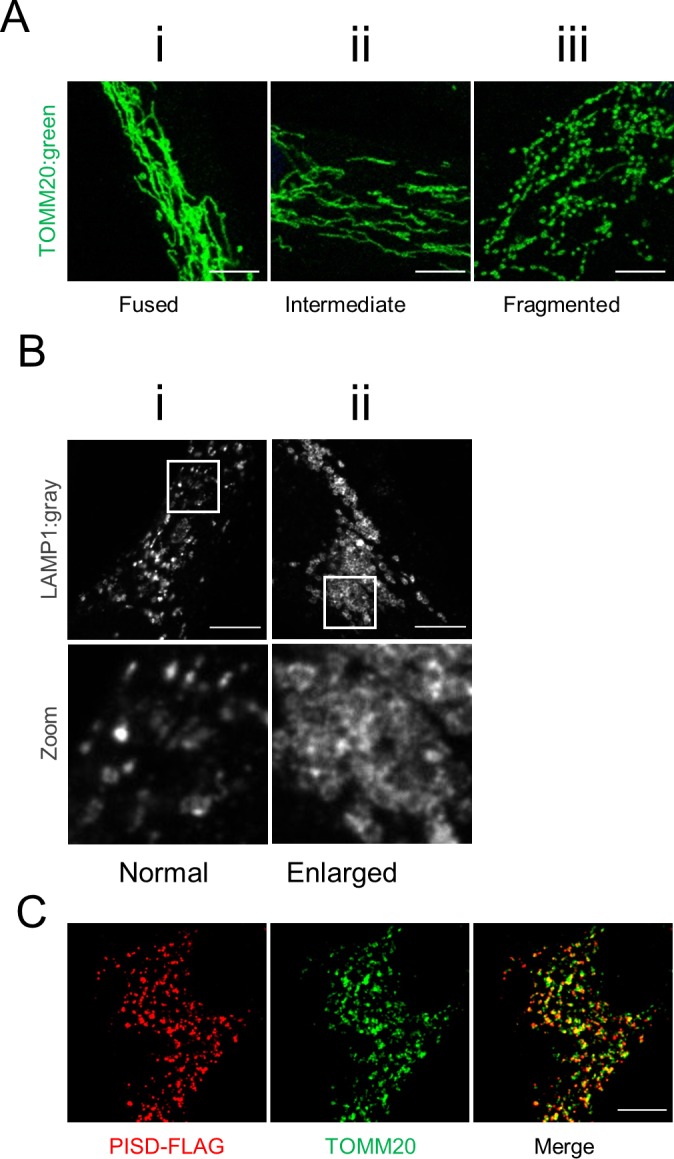
Exome sequencing of two sisters with congenital cataracts, short stature, and white matter changes identified compound heterozygous variants in the PISD gene, encoding the phosphatidylserine decarboxylase enzyme that converts phosphatidylserine to phosphatidylethanolamine (PE) in the inner mitochondrial membrane (IMM). Decreased conversion of phosphatidylserine to PE in patient fibroblasts is consistent with impaired phosphatidylserine decarboxylase (PISD) enzyme activity. Meanwhile, as evidence for mitochondrial dysfunction, patient fibroblasts exhibited more fragmented mitochondrial networks, enlarged lysosomes, decreased maximal oxygen consumption rates, and increased sensitivity to 2-deoxyglucose. Moreover, treatment with lyso-PE, which can replenish the mitochondrial pool of PE, and genetic complementation restored mitochondrial and lysosome morphology in patient fibroblasts. Functional characterization of the PISD variants demonstrates that the maternal variant causes an alternative splice product. Meanwhile, the paternal variant impairs autocatalytic self-processing of the PISD protein required for its activity. Finally, evidence for impaired activity of mitochondrial IMM proteases suggests an explanation as to why the phenotypes of these PISD patients resemble recently described "mitochondrial chaperonopathies." Collectively, these findings demonstrate that PISD is a novel mitochondrial disease gene.
© 2019 Zhao et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
-
- Beaulieu CL, Majewski J, Schwartzentruber J, Samuels ME, Fernandez BA, Bernier FP, Brudno M, Knoppers B, Marcadier J, Dyment D, et al. (2014) FORGE Canada Consortium: Outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet 94: 809–817. 10.1016/j.ajhg.2014.05.003 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases