

Coexpression patterns define epigenetic regulators associated with neurological dysfunction
- PMID: 30858344
- PMCID: PMC6442390
- DOI: 10.1101/gr.239442.118
Coexpression patterns define epigenetic regulators associated with neurological dysfunction
Abstract
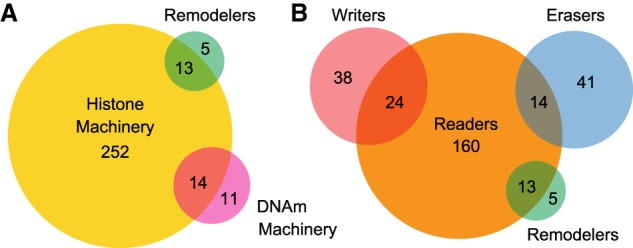
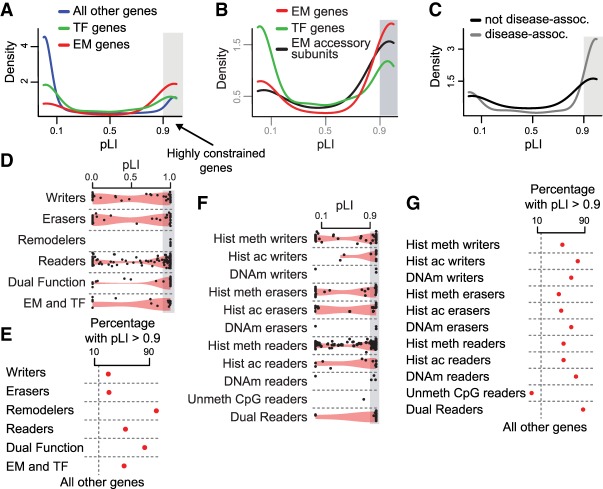
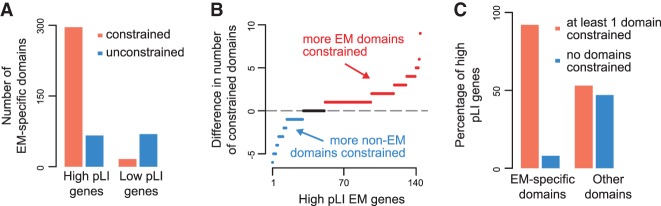
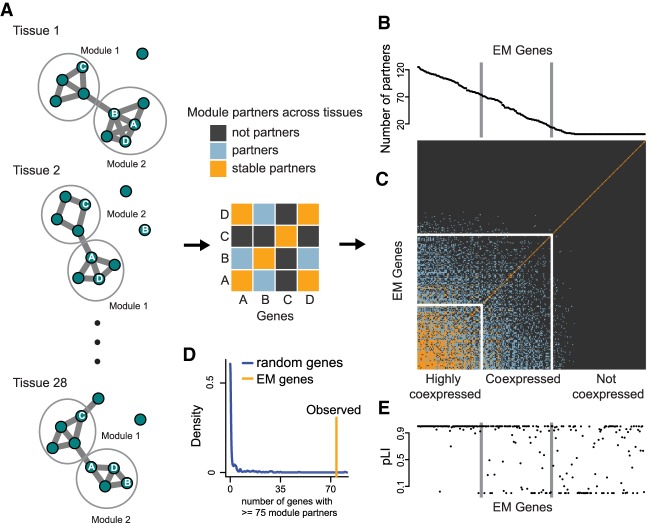
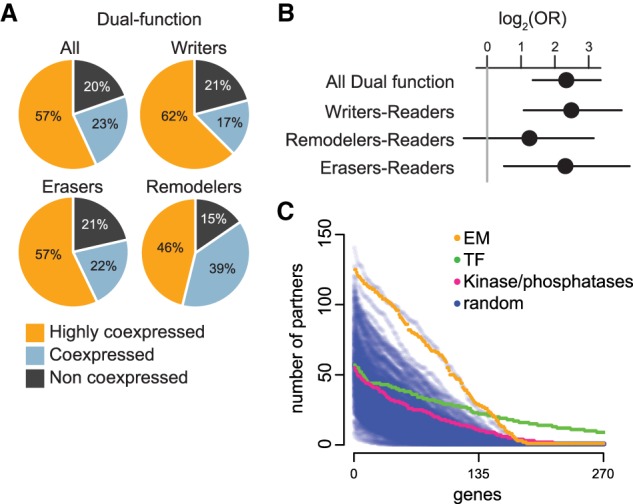
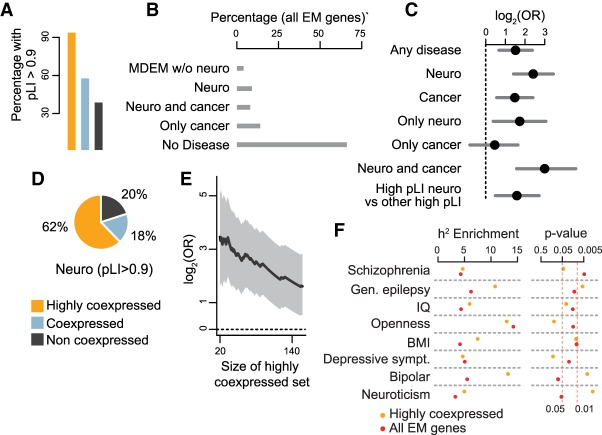
Coding variants in epigenetic regulators are emerging as causes of neurological dysfunction and cancer. However, a comprehensive effort to identify disease candidates within the human epigenetic machinery (EM) has not been performed; it is unclear whether features exist that distinguish between variation-intolerant and variation-tolerant EM genes, and between EM genes associated with neurological dysfunction versus cancer. Here, we rigorously define 295 genes with a direct role in epigenetic regulation (writers, erasers, remodelers, readers). Systematic exploration of these genes reveals that although individual enzymatic functions are always mutually exclusive, readers often also exhibit enzymatic activity (dual-function EM genes). We find that the majority of EM genes are very intolerant to loss-of-function variation, even when compared to the dosage sensitive transcription factors, and we identify 102 novel EM disease candidates. We show that this variation intolerance is driven by the protein domains encoding the epigenetic function, suggesting that disease is caused by a perturbed chromatin state. We then describe a large subset of EM genes that are coexpressed within multiple tissues. This subset is almost exclusively populated by extremely variation-intolerant genes and shows enrichment for dual-function EM genes. It is also highly enriched for genes associated with neurological dysfunction, even when accounting for dosage sensitivity, but not for cancer-associated EM genes. Finally, we show that regulatory regions near epigenetic regulators are genetically important for common neurological traits. These findings prioritize novel disease candidate EM genes and suggest that this coexpression plays a functional role in normal neurological homeostasis.
© 2019 Boukas et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N, Daly MJ, Price AL, Neale BM. 2015. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295. 10.1038/ng.3211 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical