

Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses
- PMID: 30858345
- PMCID: PMC6442391
- DOI: 10.1101/gr.240093.118
Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses
Abstract
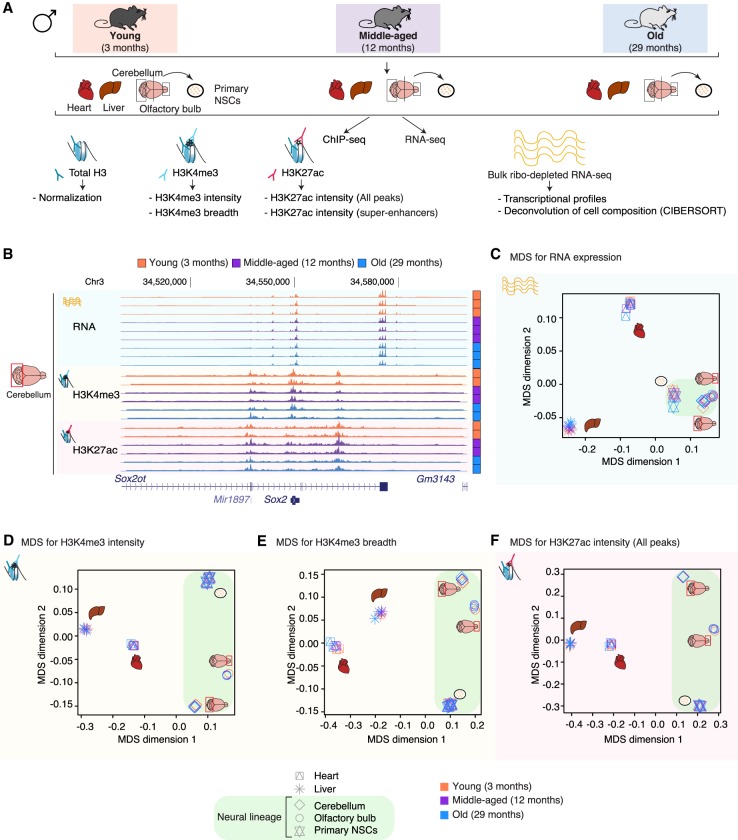
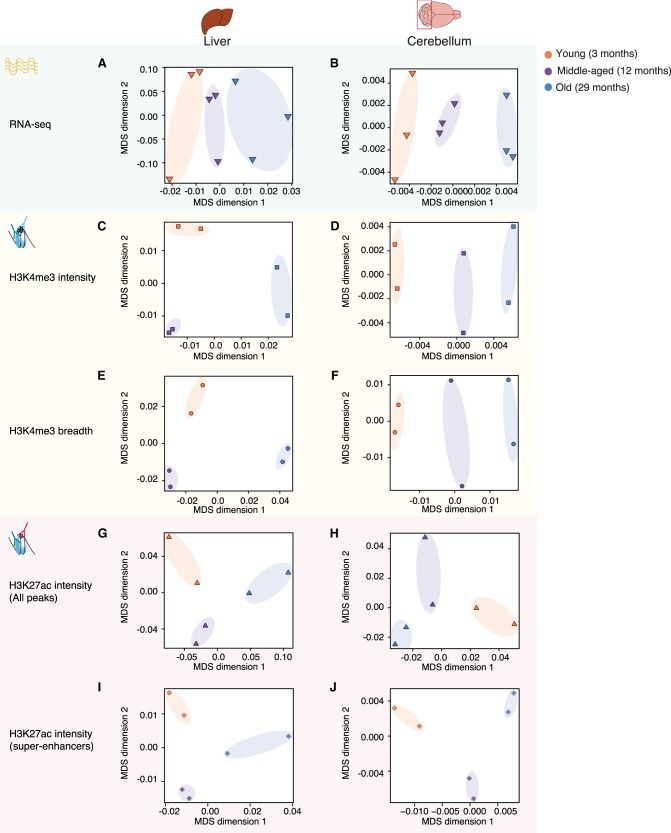
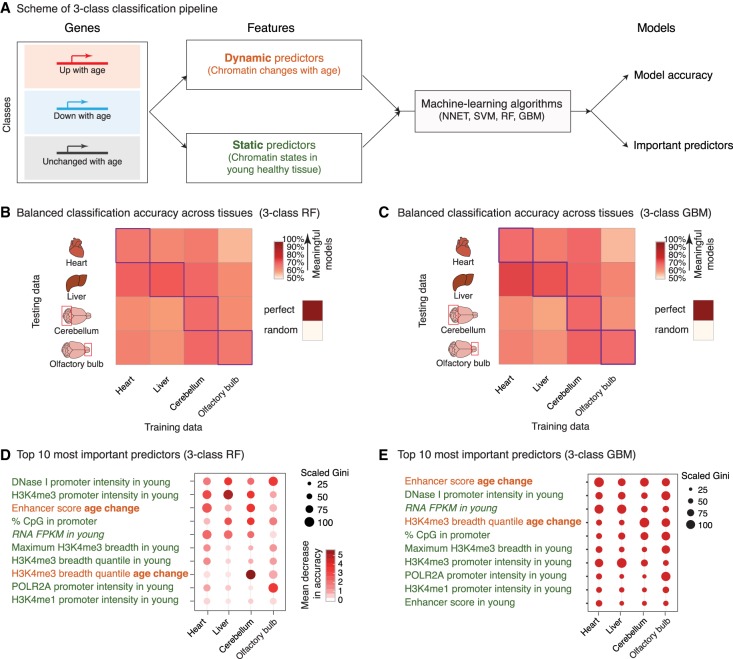
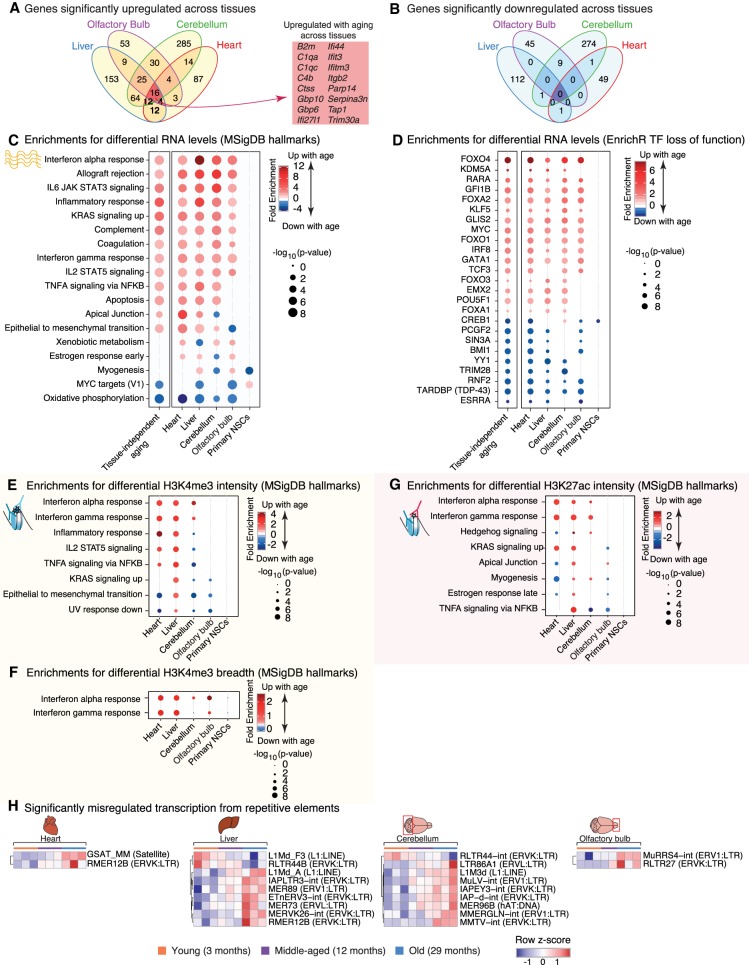
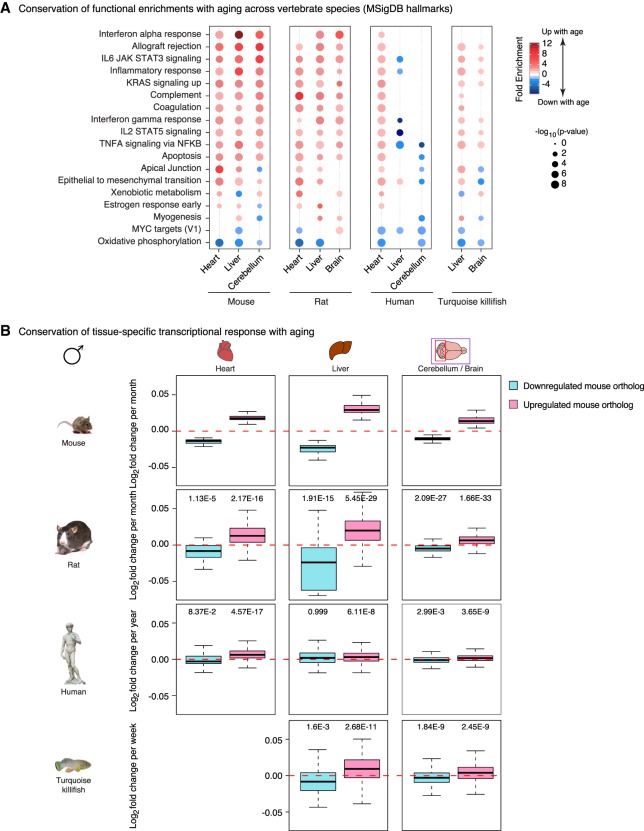
Aging is accompanied by the functional decline of tissues. However, a systematic study of epigenomic and transcriptomic changes across tissues during aging is missing. Here, we generated chromatin maps and transcriptomes from four tissues and one cell type from young, middle-aged, and old mice-yielding 143 high-quality data sets. We focused on chromatin marks linked to gene expression regulation and cell identity: histone H3 trimethylation at lysine 4 (H3K4me3), a mark enriched at promoters, and histone H3 acetylation at lysine 27 (H3K27ac), a mark enriched at active enhancers. Epigenomic and transcriptomic landscapes could easily distinguish between ages, and machine-learning analysis showed that specific epigenomic states could predict transcriptional changes during aging. Analysis of data sets from all tissues identified recurrent age-related chromatin and transcriptional changes in key processes, including the up-regulation of immune system response pathways such as the interferon response. The up-regulation of the interferon response pathway with age was accompanied by increased transcription and chromatin remodeling at specific endogenous retroviral sequences. Pathways misregulated during mouse aging across tissues, notably innate immune pathways, were also misregulated with aging in other vertebrate species-African turquoise killifish, rat, and humans-indicating common signatures of age across species. To date, our data set represents the largest multitissue epigenomic and transcriptomic data set for vertebrate aging. This resource identifies chromatin and transcriptional states that are characteristic of young tissues, which could be leveraged to restore aspects of youthful functionality to old tissues.
© 2019 Benayoun et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, et al. 2014. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346: 89–93. 10.1126/science.1252945 - DOI - PMC - PubMed
-
- Baumgart M, Groth M, Priebe S, Savino A, Testa G, Dix A, Ripa R, Spallotta F, Gaetano C, Ori M, et al. 2014. RNA-seq of the aging brain in the short-lived fish N. furzeri – conserved pathways and novel genes associated with neurogenesis. Aging Cell 13: 965–974. 10.1111/acel.12257 - DOI - PMC - PubMed
-
- Baumgart M, Priebe S, Groth M, Hartmann N, Menzel U, Pandolfini L, Koch P, Felder M, Ristow M, Englert C, et al. 2016. Longitudinal RNA-seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan. Cell Syst 2: 122–132. 10.1016/j.cels.2016.01.014 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical