

Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement
- PMID: 30858362
- PMCID: PMC6411957
- DOI: 10.1038/s41467-019-09134-9
Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement
Abstract
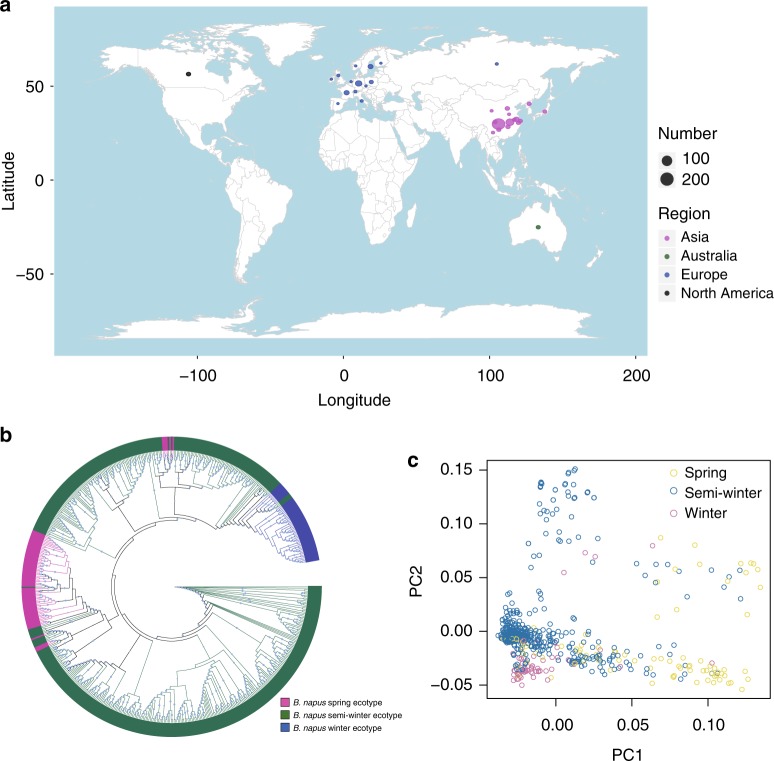
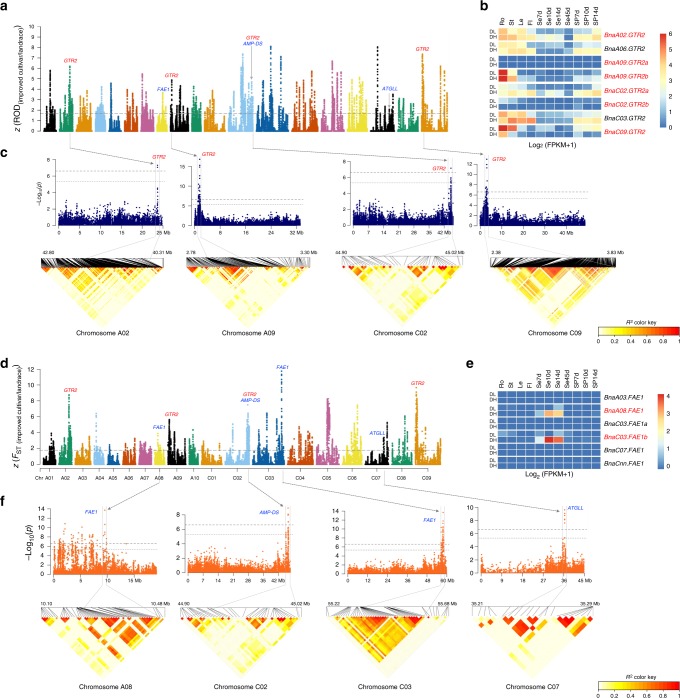
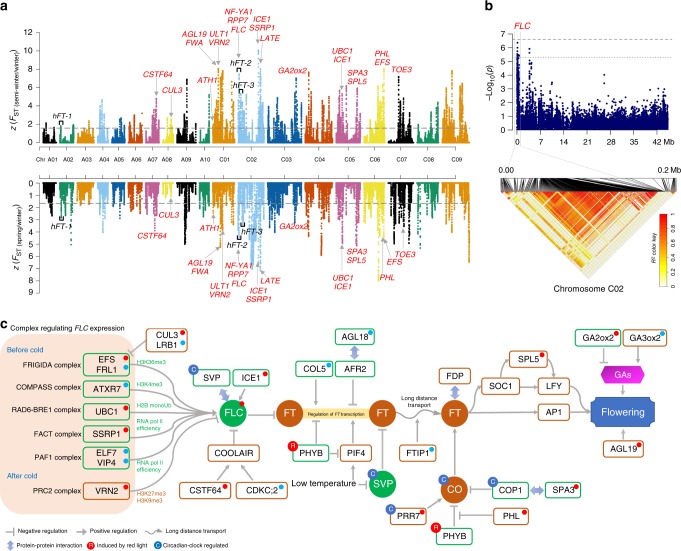
Brassica napus (2n = 4x = 38, AACC) is an important allopolyploid crop derived from interspecific crosses between Brassica rapa (2n = 2x = 20, AA) and Brassica oleracea (2n = 2x = 18, CC). However, no truly wild B. napus populations are known; its origin and improvement processes remain unclear. Here, we resequence 588 B. napus accessions. We uncover that the A subgenome may evolve from the ancestor of European turnip and the C subgenome may evolve from the common ancestor of kohlrabi, cauliflower, broccoli, and Chinese kale. Additionally, winter oilseed may be the original form of B. napus. Subgenome-specific selection of defense-response genes has contributed to environmental adaptation after formation of the species, whereas asymmetrical subgenomic selection has led to ecotype change. By integrating genome-wide association studies, selection signals, and transcriptome analyses, we identify genes associated with improved stress tolerance, oil content, seed quality, and ecotype improvement. They are candidates for further functional characterization and genetic improvement of B. napus.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Snowdon, R., Lühs, W. & Friedt, W. Oilseeds Ch. 2 (Springer, Berlin, Heidelberg, 2007).
-
- Nagaharu U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J. Bot. 1935;7:389–452.
-
- Sun F, et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017;92:452–468. - PubMed
-
- Chalhoub B, et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–953. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
