

Best practices for benchmarking germline small-variant calls in human genomes
- PMID: 30858580
- PMCID: PMC6699627
- DOI: 10.1038/s41587-019-0054-x
Best practices for benchmarking germline small-variant calls in human genomes
Erratum in
-
Author Correction: Best practices for benchmarking germline small-variant calls in human genomes.Nat Biotechnol. 2019 May;37(5):567. doi: 10.1038/s41587-019-0108-0. Nat Biotechnol. 2019. PMID: 30899106
Abstract
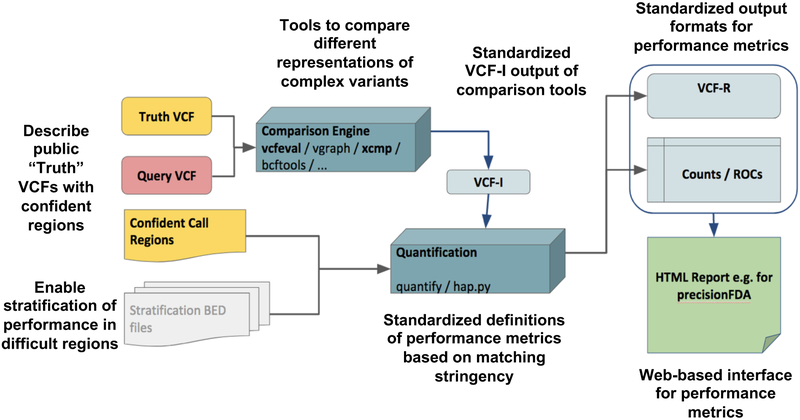
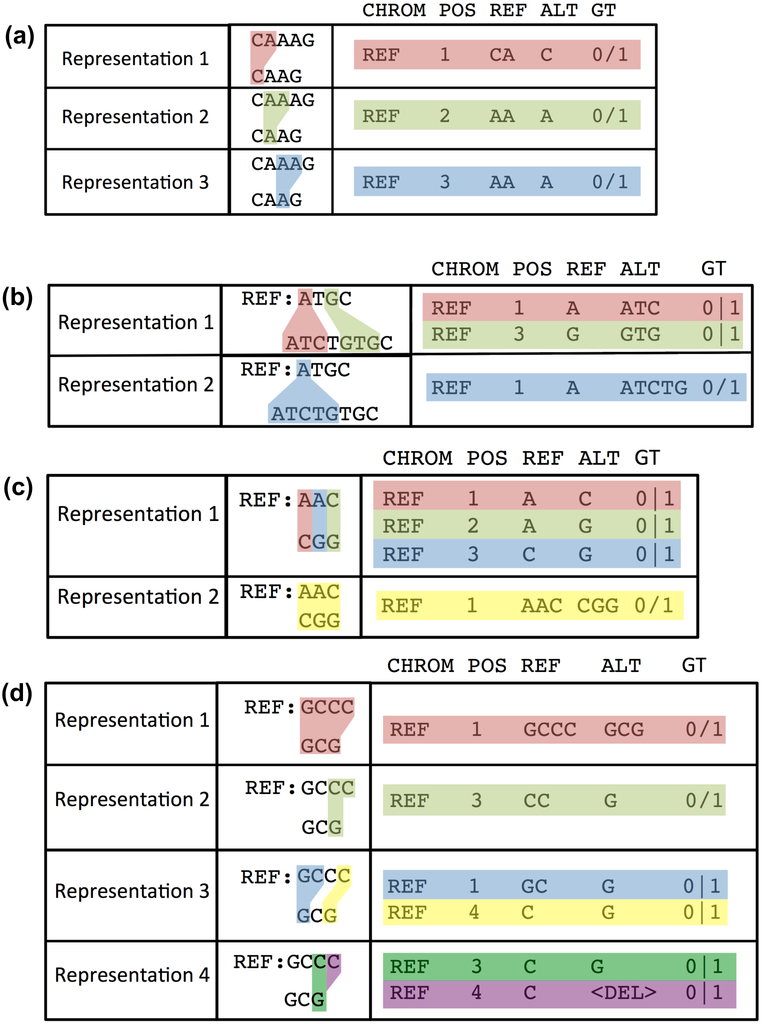
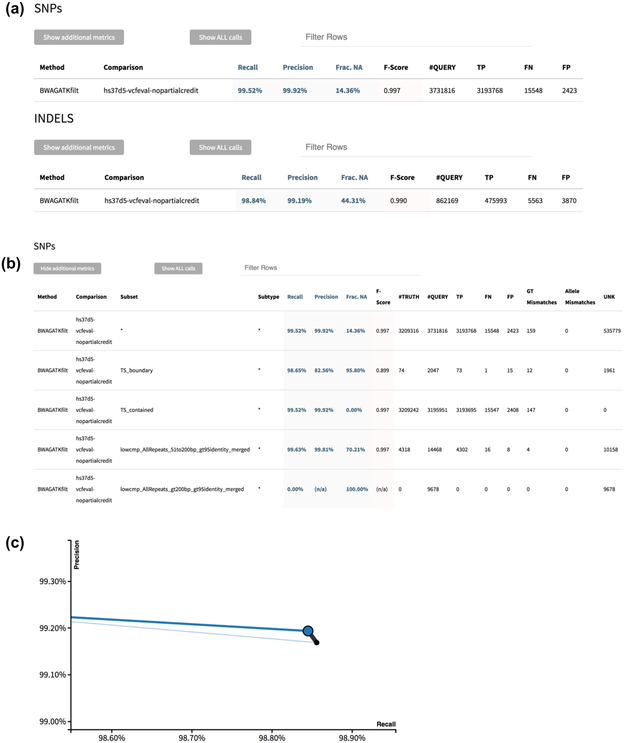
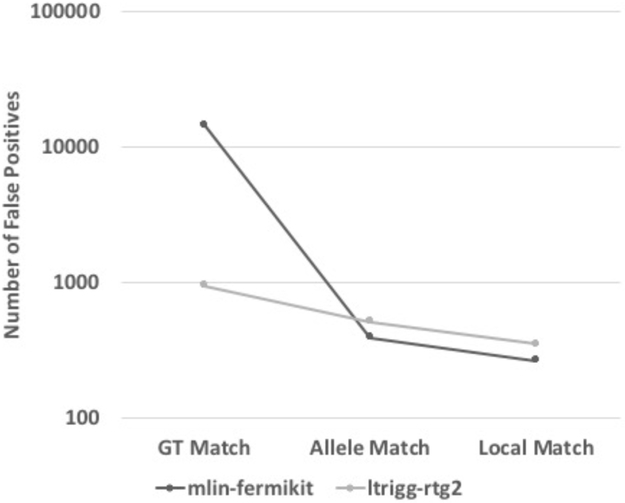
Standardized benchmarking approaches are required to assess the accuracy of variants called from sequence data. Although variant-calling tools and the metrics used to assess their performance continue to improve, important challenges remain. Here, as part of the Global Alliance for Genomics and Health (GA4GH), we present a benchmarking framework for variant calling. We provide guidance on how to match variant calls with different representations, define standard performance metrics, and stratify performance by variant type and genome context. We describe limitations of high-confidence calls and regions that can be used as truth sets (for example, single-nucleotide variant concordance of two methods is 99.7% inside versus 76.5% outside high-confidence regions). Our web-based app enables comparison of variant calls against truth sets to obtain a standardized performance report. Our approach has been piloted in the PrecisionFDA variant-calling challenges to identify the best-in-class variant-calling methods within high-confidence regions. Finally, we recommend a set of best practices for using our tools and evaluating the results.
Figures
References
-
- Xue Y, Ankala A, Wilcox WR & Hegde MR Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet. Med. 17, 444–451 (2015). - PubMed
-
- Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–51 (2014). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
