

TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons
- PMID: 30858606
- PMCID: PMC6889177
- DOI: 10.1038/s41418-019-0313-x
TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons
Abstract
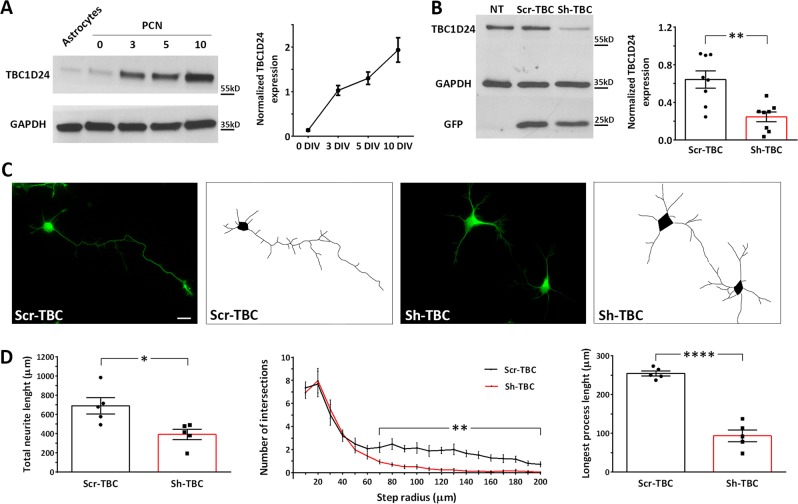
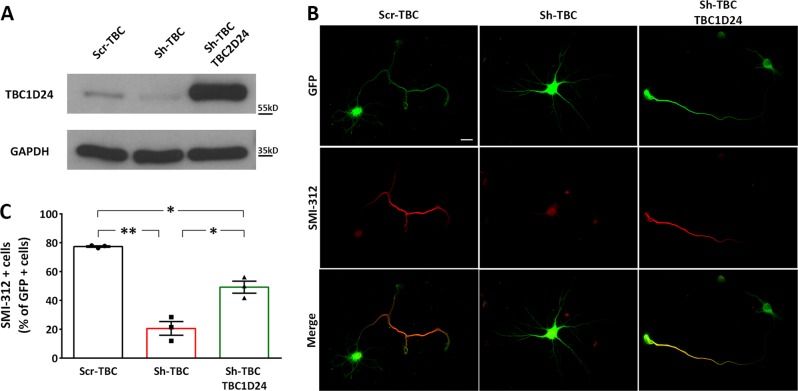
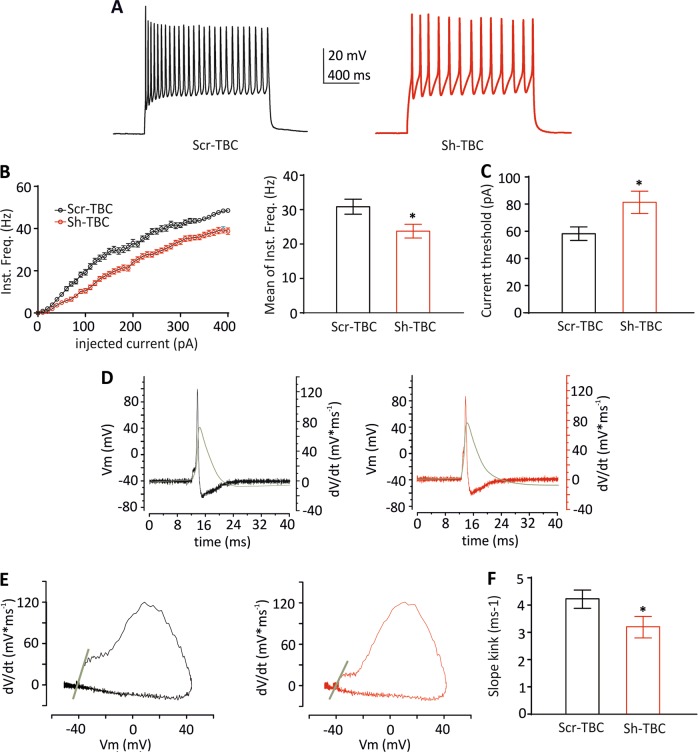
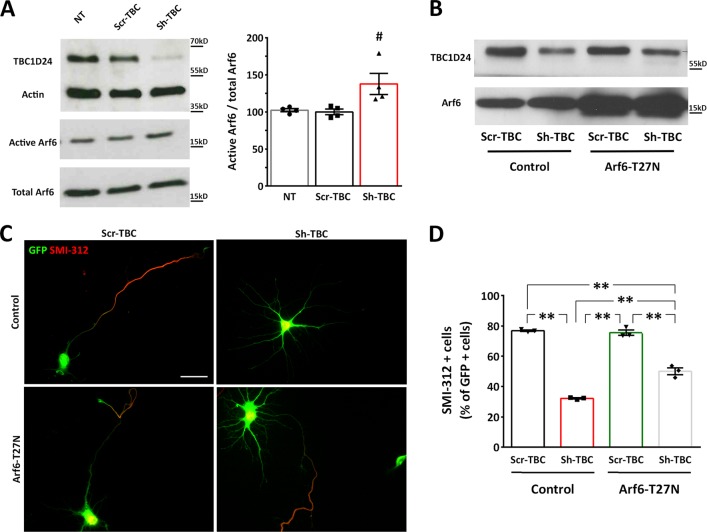
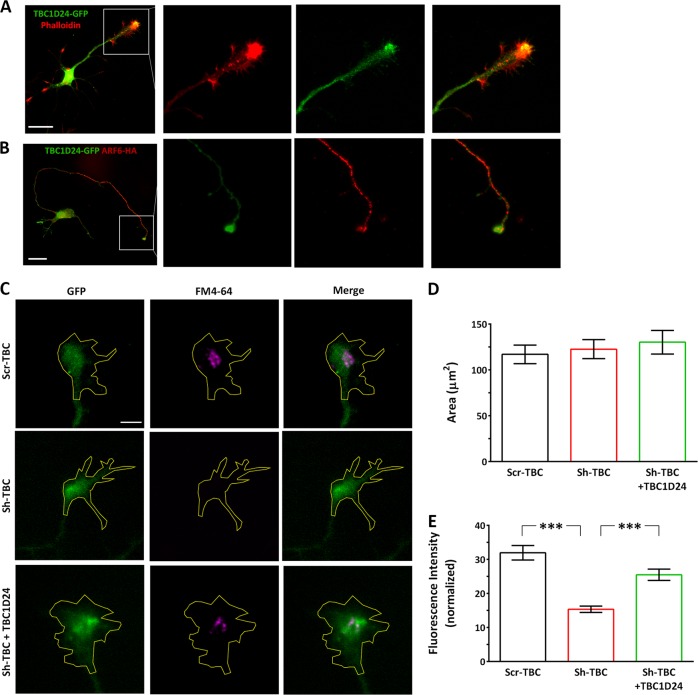
Mutations in TBC1D24 are described in patients with a spectrum of neurological diseases, including mild and severe epilepsies and complex syndromic phenotypes such as Deafness, Onycodystrophy, Osteodystrophy, Mental Retardation and Seizure (DOORS) syndrome. The product of TBC1D24 is a multifunctional protein involved in neuronal development, regulation of synaptic vesicle trafficking, and protection from oxidative stress. Although pathogenic mutations in TBC1D24 span the entire coding sequence, no clear genotype/phenotype correlations have emerged. However most patients bearing predicted loss of function mutations exhibit a severe neurodevelopmental disorder. Aim of the study is to investigate the impact of TBC1D24 knockdown during the first stages of neuronal differentiation when axonal specification and outgrowth take place. In rat cortical primary neurons silenced for TBC1D24, we found defects in axonal specification, the maturation of axonal initial segment and action potential firing. The axonal phenotype was accompanied by an impairment of endocytosis at the growth cone and an altered activation of the TBC1D24 molecular partner ADP ribosylation factor 6. Accordingly, acute knockdown of TBC1D24 in cerebrocortical neurons in vivo analogously impairs callosal projections. The axonal defect was also investigated in human induced pluripotent stem cell-derived neurons from patients carrying TBC1D24 mutations. Reprogrammed neurons from a patient with severe developmental encephalopathy show significant axon formation defect that were absent from reprogrammed neurons of a patient with mild early onset epilepsy. Our data reveal that alterations of membrane trafficking at the growth cone induced by TBC1D24 loss of function cause axonal and excitability defects. The axonal phenotype correlates with the disease severity and highlight an important role for TBC1D24 in connectivity during brain development.
Conflict of interest statement
The authors declare no conflict of interests.
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
