

Erythromycin acts through the ghrelin receptor to attenuate inflammatory responses in chondrocytes and maintain joint integrity
- PMID: 30862504
- PMCID: PMC7704045
- DOI: 10.1016/j.bcp.2019.03.014
Erythromycin acts through the ghrelin receptor to attenuate inflammatory responses in chondrocytes and maintain joint integrity
Abstract
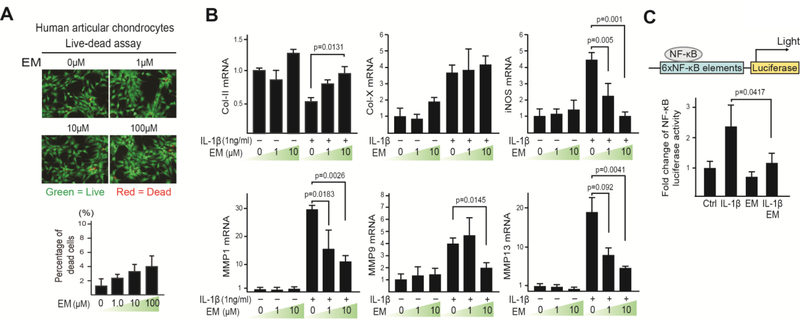
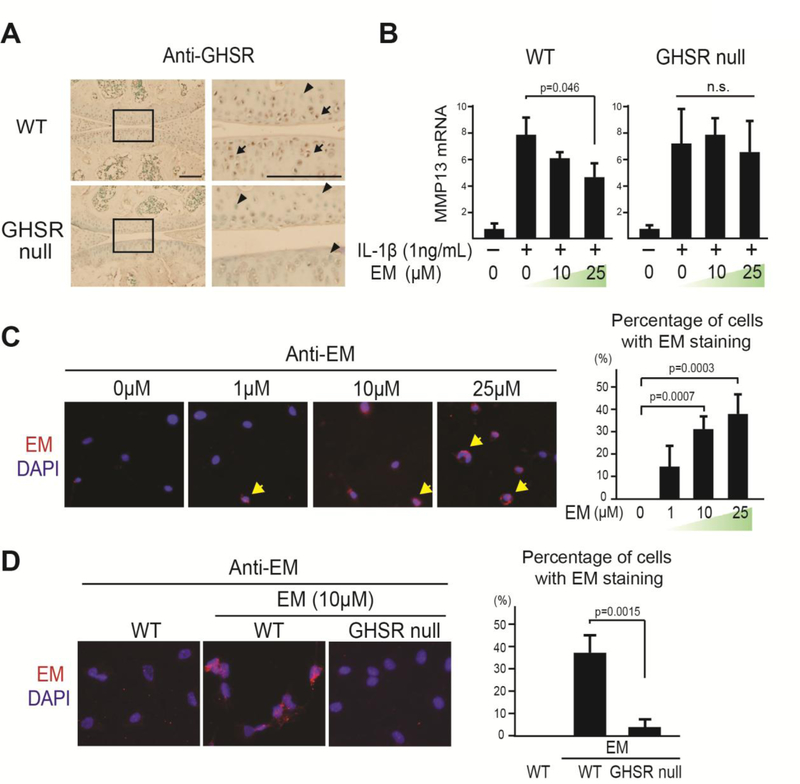
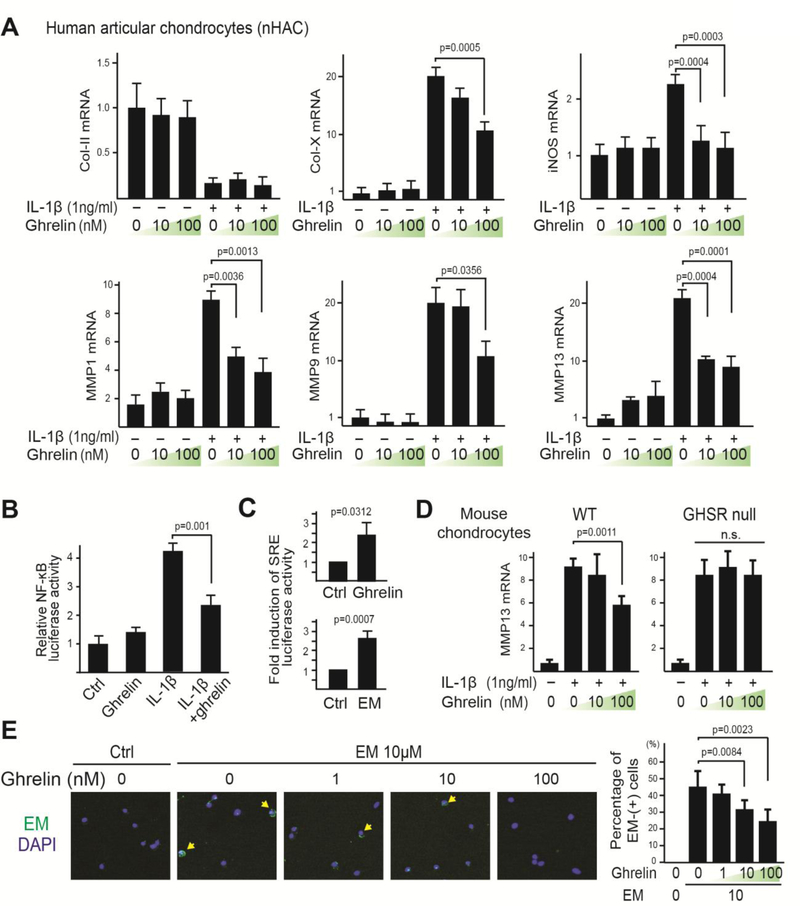
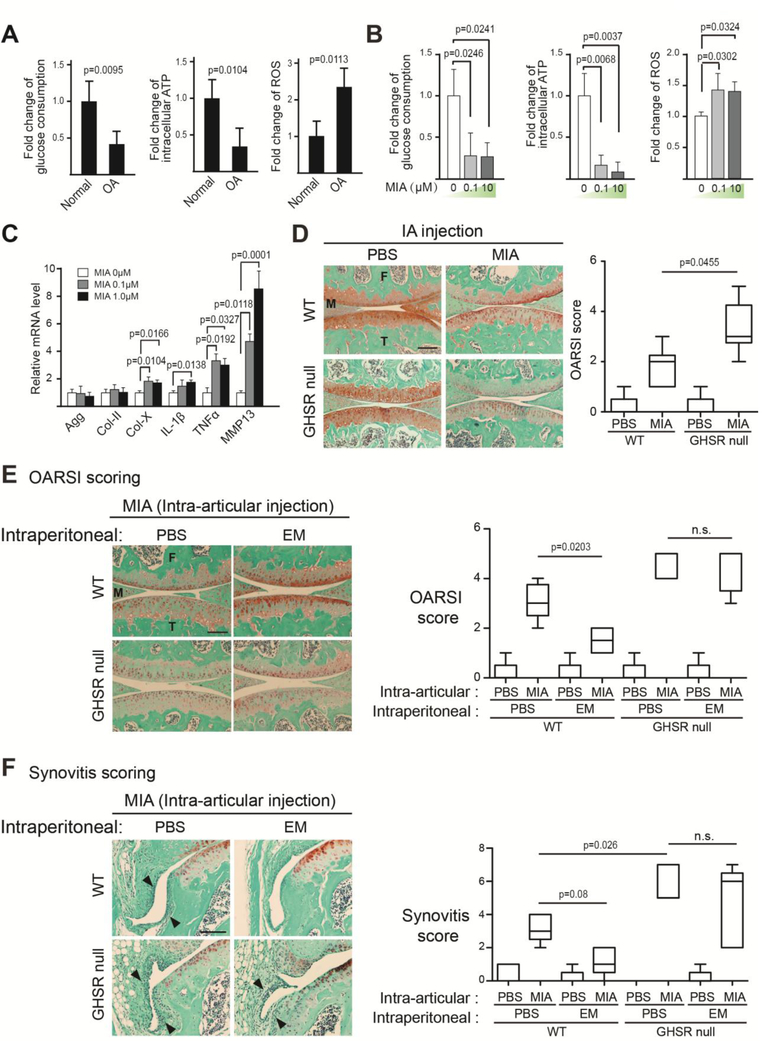
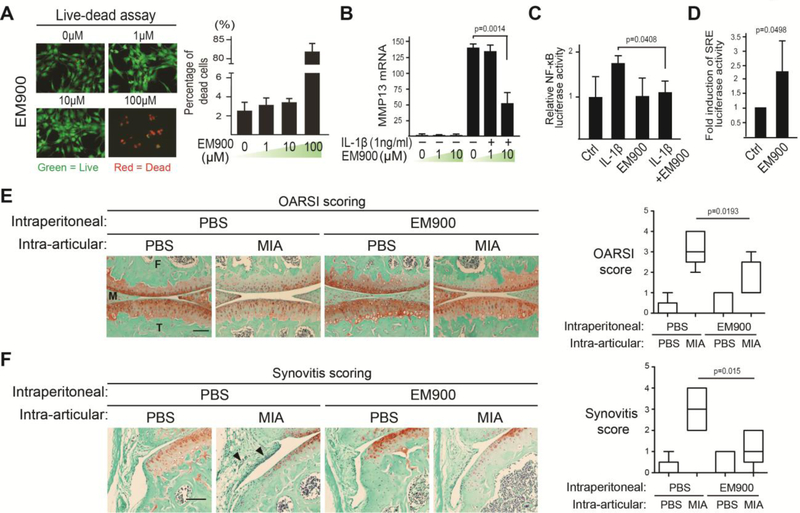
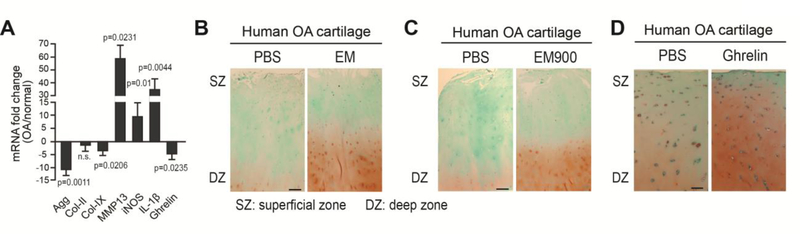
Osteoarthritis (OA) is a prevalent disease characterized by chronic joint degeneration and low-grade localized inflammation. There is no available treatment to delay OA progression. We report that in human primary articular chondrocytes, erythromycin, a well-known macrolide antibiotic, had the ability to inhibit pro-inflammatory cytokine Interleukin 1β (IL-1β)-induced catabolic gene expression and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation. Furthermore, erythromycin inhibited monosodium iodoacetate (MIA)-induced joint inflammation and cartilage matrix destruction in mice, an arthritis model that reflects the inflammatory and cartilage matrix loss aspects of OA. EM900, an erythromycin-derivative lacking antibiotic function, had the same activity as erythromycin in vitro and in vivo, indicating distinct anti-inflammatory and antibiotic properties. Using an antibody against erythromycin, we found erythromycin was present on chondrocytes in a dose-dependent manner. The association of erythromycin with chondrocytes was diminished in ghrelin receptor null chondrocytes, and administration of the ghrelin ligand prevented the association of erythromycin with chondrocytes. Importantly, the anti-inflammatory activity of erythromycin was diminished in ghrelin receptor null chondrocytes. Moreover, erythromycin could not exert its chondroprotective effect in ghrelin receptor null mice, and the loss of ghrelin receptor further augmented joint damage upon MIA-injection. Therefore, our study identified a novel pharmacological mechanism for how erythromycin exerts its chondroprotective effect. This mechanism entails ghrelin receptor signaling, which is necessary for alleviating inflammation and joint destruction.
Keywords: Cartilage; Chondrocyte; Erythromycin; Ghrelin receptor; Osteoarthritis; Synovitis.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest
None of the authors have any conflict of interest related to this work.
Figures
References
-
- Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation --- United States, 2007–2009, MMWR Morb Mortal Wkly Rep, 59 (2010) 1261–1265. - PubMed
-
- Bitton R, The economic burden of osteoarthritis, Am J Manag Care, 15 (2009) S230–235. - PubMed
-
- Hochberg MC, Osteoarthritis year 2012 in review: clinical, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 20 (2012) 1465–1469. - PubMed
-
- Lories RJ, Luyten FP, The bone-cartilage unit in osteoarthritis, Nature reviews. Rheumatology, 7 (2011) 43–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
