

How I manage patients with Wiskott Aldrich syndrome
- PMID: 30864154
- PMCID: PMC7612067
- DOI: 10.1111/bjh.15831
How I manage patients with Wiskott Aldrich syndrome
Abstract
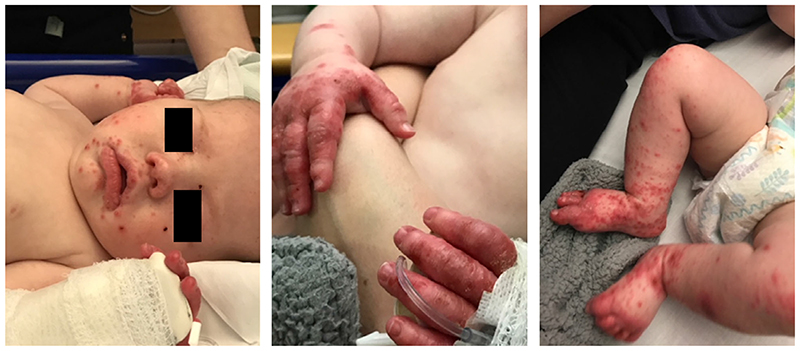
Wiskott Aldrich syndrome (WAS) is a primary immunodeficiency disease resulting in recurrent infections, eczema and microthrombocytopaenia. In its classical form, significant combined immune deficiency, autoimmune complications and risk of haematological malignancy necessitate early correction with stem cell transplantation or gene therapy. A milder form, X-linked thrombocytopaenia (XLT), shares similar bleeding risk from thrombocytopaenia but is not associated with other significant clinical features and is generally managed conservatively. Here, we detail our approach to the diagnosis and treatment of classical WAS and XLT.
Keywords: Wiskott Aldrich syndrome; X-linked thrombocytopenia; haematopoietic stem cell transplant; immunodeficiency.
© 2019 British Society for Haematology and John Wiley & Sons Ltd.
Figures

References
-
- Albert MH, Bittner TC, Nonoyama S, Notaran-gelo LD, Burns S, Imai K, Espanol T, Fasth A, Pellier I, Strauss G, Morio T, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115:3231–3238. - PubMed
-
- Andres O, Henning K, Strauss G, Pflug A, Manukjan G, Schulze H. Diagnosis of platelet function disorders: a standardized, rational, and modular flow cytometric approach. Platelets. 2018;29:347–356. - PubMed
-
- Andreu N, Pujol-Moix N, Martinez-Lostao L, Oset M, Muniz-Diaz E, Estivill X, Volpini V, Fillat C. Wiskott-Aldrich syndrome in a female with skewed X-chromosome inactivation. Blood Cells, Molecules, & Diseases. 2003;31:332–337. - PubMed
-
- Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, Modlich U, Beier R, Gohring G, Steinemann D, Fronza R, et al. Gene therapy for Wiskott-Aldrich syndrome-long-term efficacy and genotoxicity. Science Translational Medicine. 2014;6:227ra233 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
