

Stretching magnitude-dependent inactivation of AKT by ROS led to enhanced p53 mitochondrial translocation and myoblast apoptosis
- PMID: 30865562
- PMCID: PMC6724521
- DOI: 10.1091/mbc.E18-12-0770
Stretching magnitude-dependent inactivation of AKT by ROS led to enhanced p53 mitochondrial translocation and myoblast apoptosis
Abstract
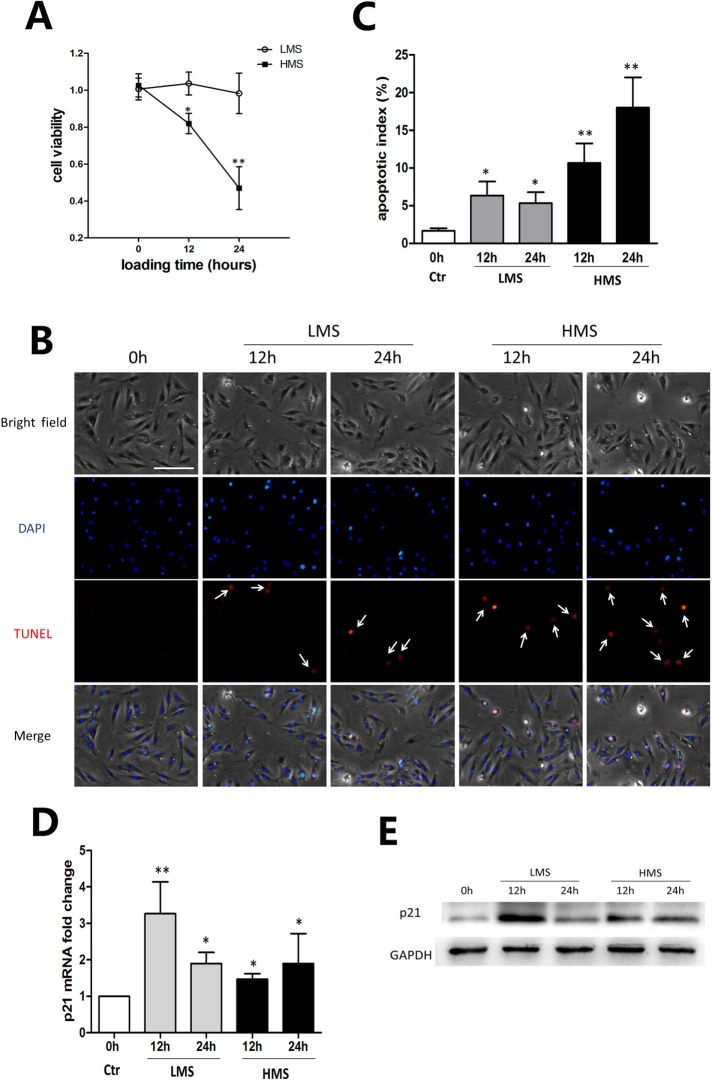
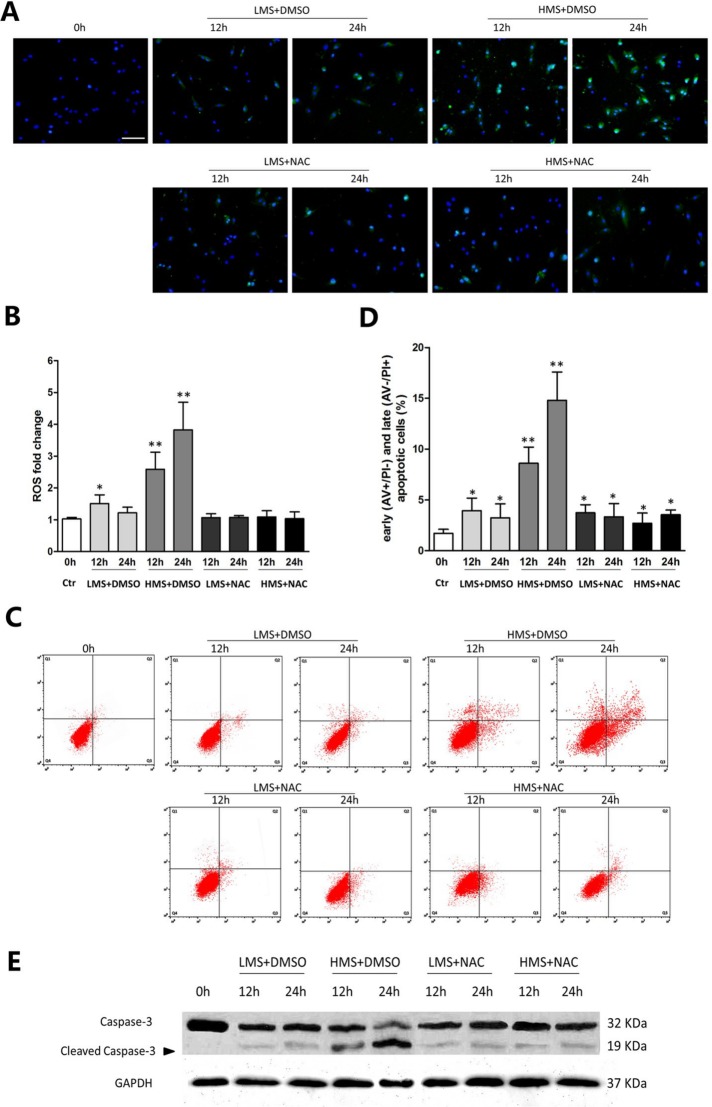
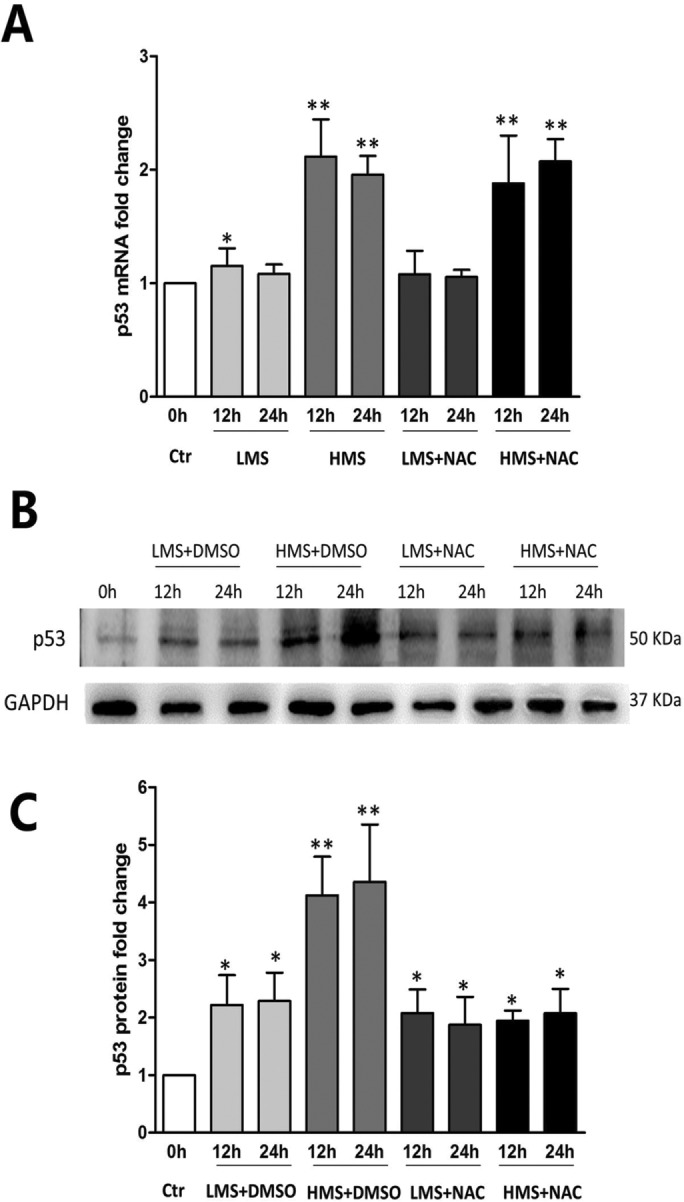
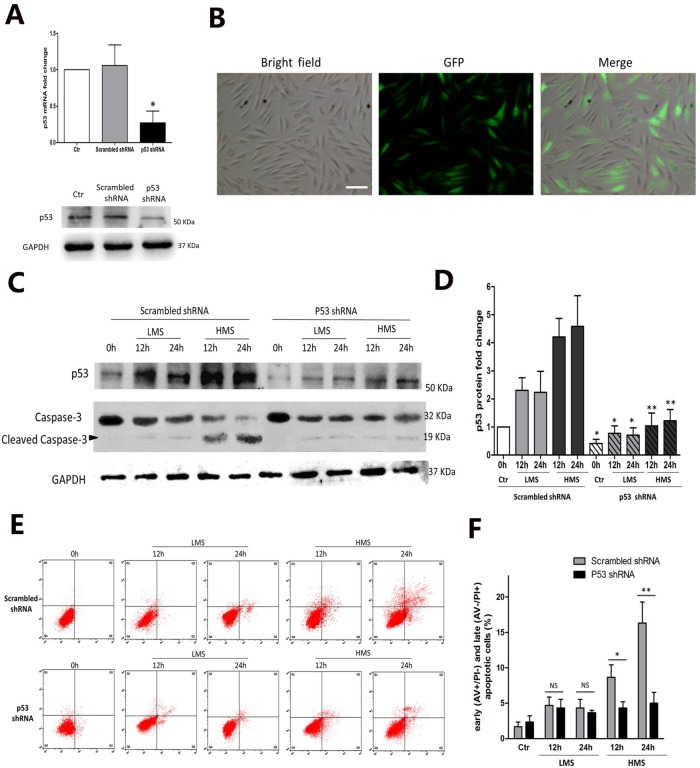
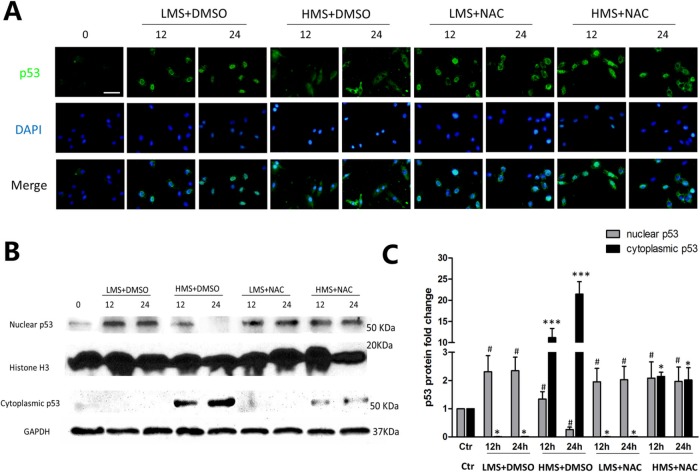
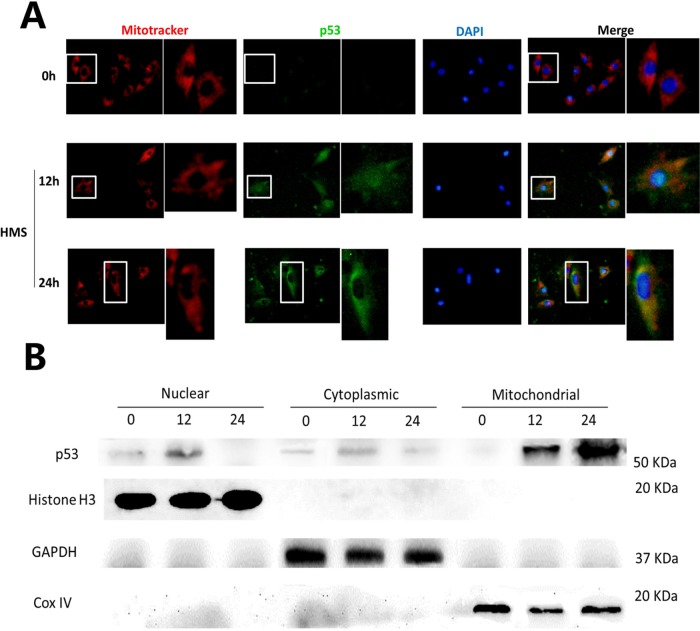
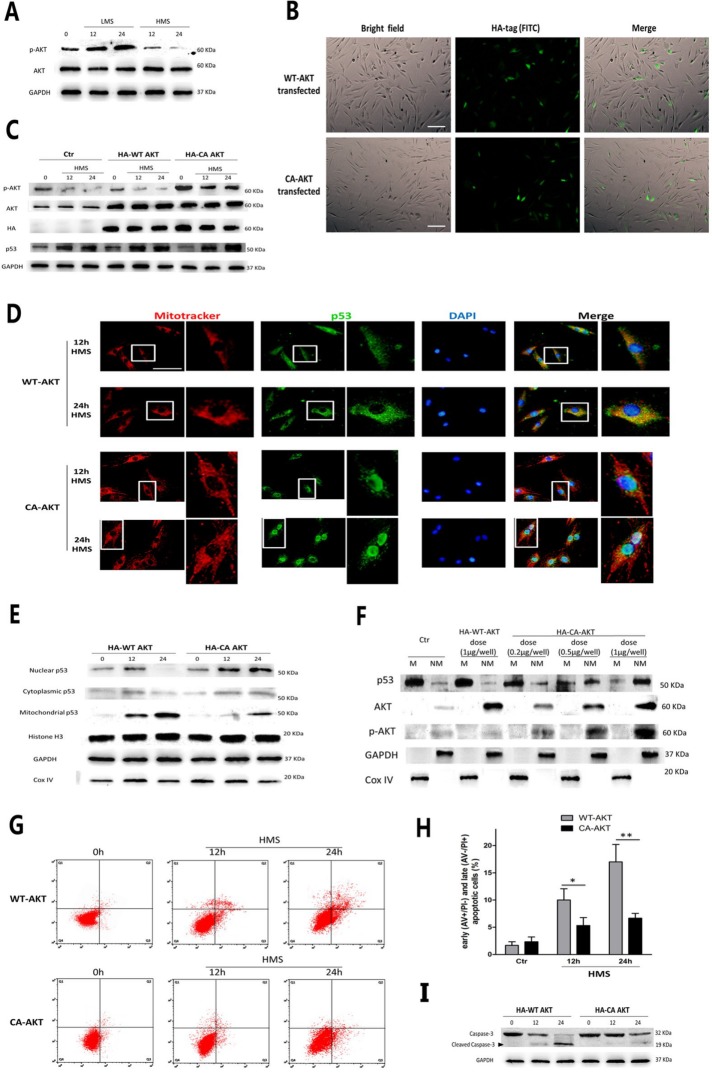
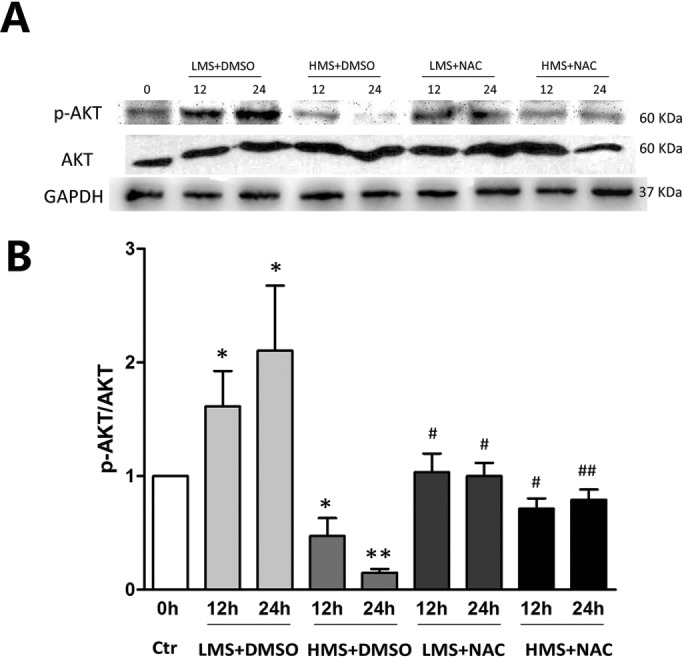
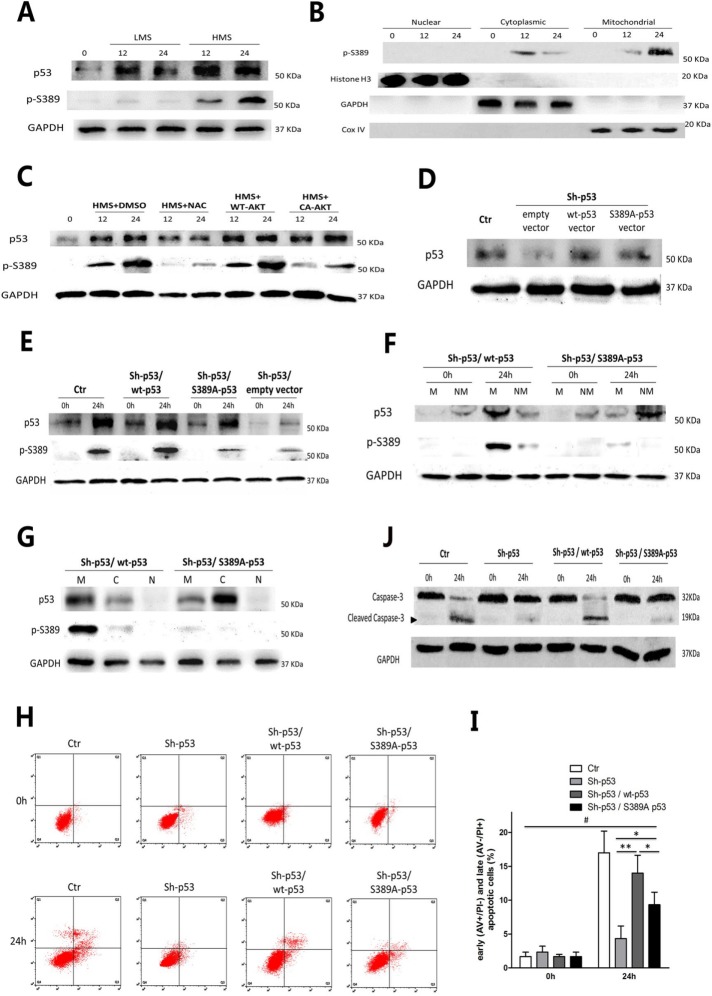
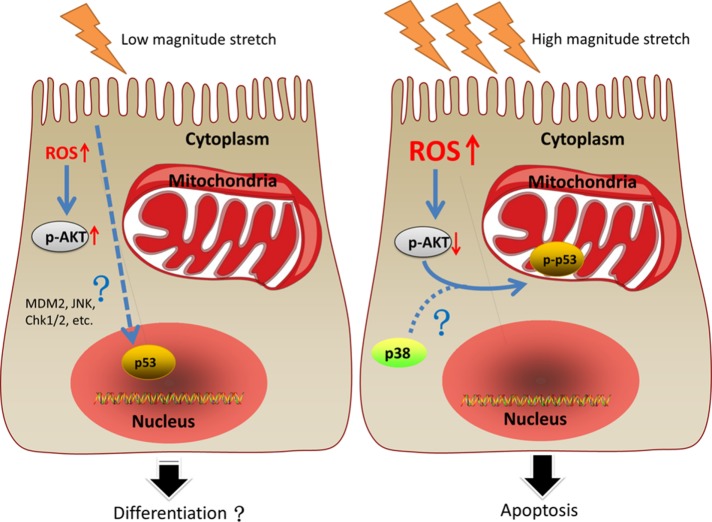
Previously, we had shown that high magnitude stretch (HMS), rather than low magnitude stretch (LMS), induced significant apoptosis of skeletal muscle C2C12 myoblasts. However, the molecular mechanism remains obscure. In this study, we found that p53 protein accumulated in the nucleus of LMS-loaded cells, whereas it translocated into mitochondria of HMS-loaded cells. Knocking down endogenous p53 by shRNA abrogated HMS-induced apoptosis. Furthermore, we demonstrated that overaccumulation of reactive oxygen species (ROS) during HMS-inactivated AKT that was activated in LMS-treated cells, which accounted for the distinct p53 subcellular localizations under HMS and LMS. Blocking ROS generation by N-acetylcysteine (NAC) or overexpressing constitutively active AKT vector (CA-AKT) inhibited HMS-incurred p53 mitochondrial translocation and promoted its nuclear targeting. Moreover, both NAC and CA-AKT significantly attenuated HMS-induced C2C12 apoptosis. Finally, we found that Ser389 phosphorylation of p53 was a downstream event of ROS-inactivated AKT pathway, which was critical to p53 mitochondrial trafficking during HMS stimuli. Transfecting p53-shRNA C2C12s with the mutant p53 (S389A) that was unable to target p53 to mitochondria underwent significantly lower apoptosis than transfection with wild-type p53. Altogether, our study uncovered that mitochondrial localization of p53, resulting from p53 Ser389 phosphorylation through ROS-inactivated AKT pathway, prompted C2C12 myoblast apoptosis during HMS stimulation.
Figures
Similar articles
-
Protein tyrosine phosphatase 1B inhibition ameliorates palmitate-induced mitochondrial dysfunction and apoptosis in skeletal muscle cells.Free Radic Biol Med. 2013 Dec;65:1435-1446. doi: 10.1016/j.freeradbiomed.2013.09.019. Epub 2013 Oct 10. Free Radic Biol Med. 2013. PMID: 24120971
-
Involvement of Bcl-2 family members, phosphatidylinositol 3'-kinase/AKT and mitochondrial p53 in curcumin (diferulolylmethane)-induced apoptosis in prostate cancer.Int J Oncol. 2007 Apr;30(4):905-18. Int J Oncol. 2007. PMID: 17332930
-
Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway.Arch Toxicol. 2012 Jun;86(6):923-33. doi: 10.1007/s00204-012-0864-9. Epub 2012 May 24. Arch Toxicol. 2012. PMID: 22622864
-
[Icariin reduces S-nitrosogultathione induced endothelial cell apoptosis through modulating AKT/P53 pathway].Zhonghua Xin Xue Guan Bing Za Zhi. 2016 Aug 24;44(8):707-13. doi: 10.3760/cma.j.issn.0253-3758.2016.08.013. Zhonghua Xin Xue Guan Bing Za Zhi. 2016. PMID: 27545131 Chinese.
-
Potential enhancement of post-stroke angiogenic response by targeting the oligomeric aggregation of p53 protein.Front Cell Neurosci. 2023 Jul 18;17:1193362. doi: 10.3389/fncel.2023.1193362. eCollection 2023. Front Cell Neurosci. 2023. PMID: 37534043 Free PMC article. Review.
Cited by
-
Autophagy in muscle regeneration: potential therapies for myopathies.J Cachexia Sarcopenia Muscle. 2022 Jun;13(3):1673-1685. doi: 10.1002/jcsm.13000. Epub 2022 Apr 17. J Cachexia Sarcopenia Muscle. 2022. PMID: 35434959 Free PMC article. Review.
-
Intensive stretch-activated CRT-PMCA1 feedback loop promoted apoptosis of myoblasts through Ca2+ overloading.Apoptosis. 2022 Dec;27(11-12):929-945. doi: 10.1007/s10495-022-01759-4. Epub 2022 Aug 17. Apoptosis. 2022. PMID: 35976579
-
Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury.Molecules. 2020 Jul 17;25(14):3267. doi: 10.3390/molecules25143267. Molecules. 2020. PMID: 32709024 Free PMC article.
-
P53 Regulates the Redox Status of Lung Endothelial Cells.Inflammation. 2020 Apr;43(2):686-691. doi: 10.1007/s10753-019-01150-7. Inflammation. 2020. PMID: 31838664 Free PMC article.
-
N-acetylcysteine promotes cyclic mechanical stress-induced osteogenic differentiation of periodontal ligament stem cells by down-regulating Nrf2 expression.J Dent Sci. 2022 Apr;17(2):750-762. doi: 10.1016/j.jds.2021.10.003. Epub 2021 Oct 28. J Dent Sci. 2022. PMID: 35756790 Free PMC article.
References
-
- Abraham AG, O’Neill E. (2014). PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans , 798–803. - PubMed
-
- Akimoto T, Ushida T, Miyaki S, Tateishi T, Fukubayashi T. (2001). Mechanical stretch is a down-regulatory signal for differentiation of C2C12. Mater Sci Eng C , 75–78.
-
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. (2010). Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol , 83–91. - PubMed
-
- Andersen JI, Pennisi CP, Fink T, Zachar V. (2018). Focal adhesion kinase activation is necessary for stretch-induced alignment and enhanced differentiation of myogenic precursor cells. Tissue Eng Part A , 631–640. - PubMed
-
- Appella E, Anderson CW. (2001). Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem , 2764–2772. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
