

The post-cardiac arrest syndrome: A case for lung-brain coupling and opportunities for neuroprotection
- PMID: 30866740
- PMCID: PMC6547189
- DOI: 10.1177/0271678X19835552
The post-cardiac arrest syndrome: A case for lung-brain coupling and opportunities for neuroprotection
Abstract
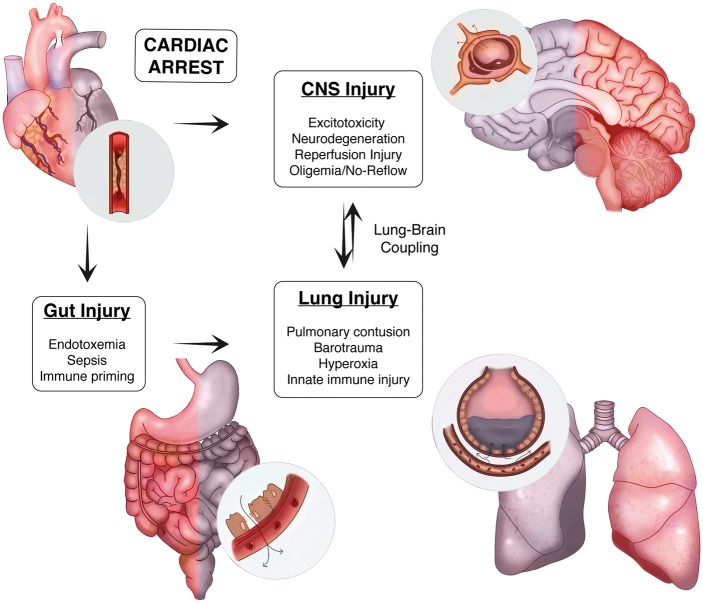
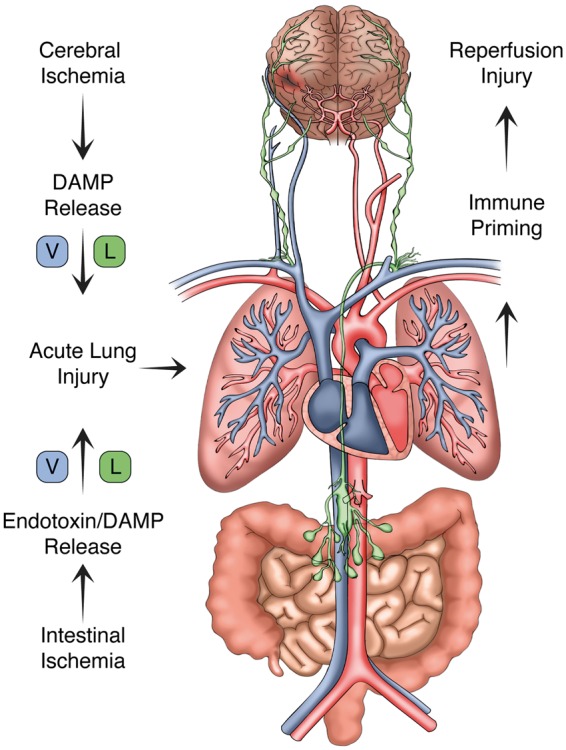
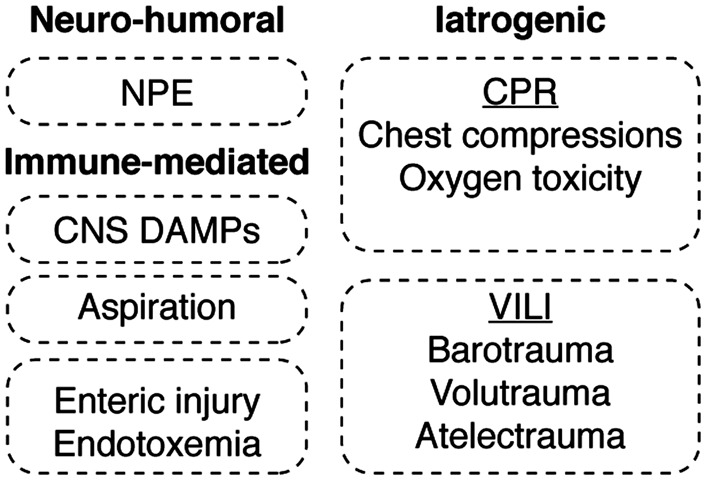
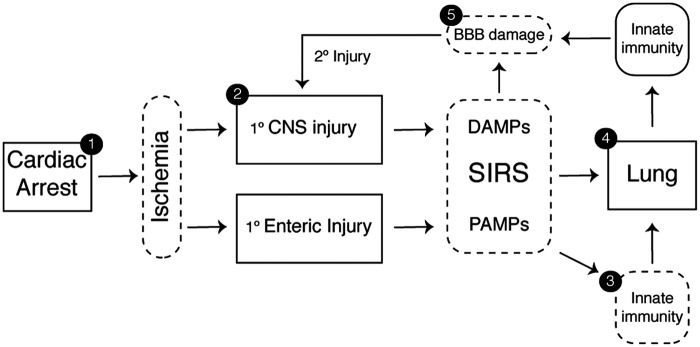
Systemic inflammation and multi-organ failure represent hallmarks of the post-cardiac arrest syndrome (PCAS) and predict severe neurological injury and often fatal outcomes. Current interventions for cardiac arrest focus on the reversal of precipitating cardiac pathologies and the implementation of supportive measures with the goal of limiting damage to at-risk tissue. Despite the widespread use of targeted temperature management, there remain no proven approaches to manage reperfusion injury in the period following the return of spontaneous circulation. Recent evidence has implicated the lung as a moderator of systemic inflammation following remote somatic injury in part through effects on innate immune priming. In this review, we explore concepts related to lung-dependent innate immune priming and its potential role in PCAS. Specifically, we propose and investigate the conceptual model of lung-brain coupling drawing from the broader literature connecting tissue damage and acute lung injury with cerebral reperfusion injury. Subsequently, we consider the role that interventions designed to short-circuit lung-dependent immune priming might play in improving patient outcomes following cardiac arrest and possibly other acute neurological injuries.
Keywords: Cardiac arrest; acute lung injury; blood–brain barrier; damage-associated molecular patterns; innate immune priming; ischemic neurodegeneration; neuroprotection; neutrophil; pathogen-associated molecular patterns; post-cardiac arrest syndrome; sepsis; systemic inflammatory response syndrome.
Figures
References
-
- Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA 2010; 303: 2165–2171. - PubMed
-
- Albert CM, Mittleman MA, Chae CU, et al. Triggering of sudden death from cardiac causes by vigorous exertion. N Engl J Med 2000; 343: 1355–1361. - PubMed
-
- Wood MA, Stambler BS, Damiano RJ, et al. Lessons learned from data logging in a multicenter clinical trial using a late-generation implantable cardioverter-defibrillator. The Guardian ATP 4210 Multicenter Investigators Group. J Am Coll Cardiol 1994; 24: 1692–1699. - PubMed
-
- Nadkarni VM, Larkin GL, Peberdy MA, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA 2006; 295: 50–57. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
