

RBFox2-miR-34a-Jph2 axis contributes to cardiac decompensation during heart failure
- PMID: 30867288
- PMCID: PMC6442575
- DOI: 10.1073/pnas.1822176116
RBFox2-miR-34a-Jph2 axis contributes to cardiac decompensation during heart failure
Abstract
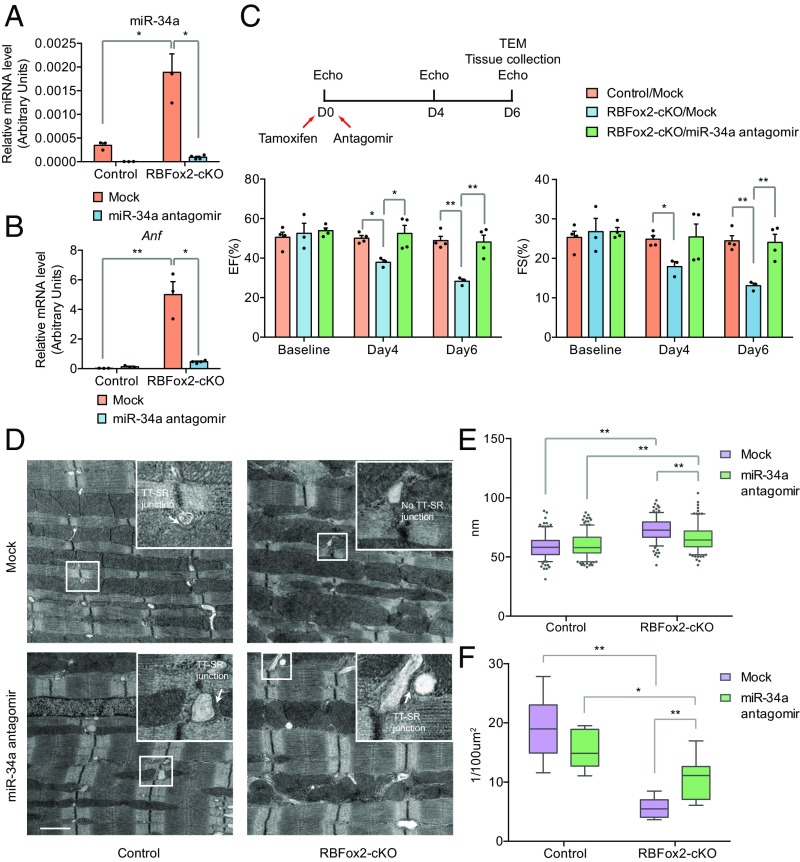
Heart performance relies on highly coordinated excitation-contraction (EC) coupling, and defects in this critical process may be exacerbated by additional genetic defects and/or environmental insults to cause eventual heart failure. Here we report a regulatory pathway consisting of the RNA binding protein RBFox2, a stress-induced microRNA miR-34a, and the essential EC coupler JPH2. In this pathway, initial cardiac defects diminish RBFox2 expression, which induces transcriptional repression of miR-34a, and elevated miR-34a targets Jph2 to impair EC coupling, which further manifests heart dysfunction, leading to progressive heart failure. The key contribution of miR-34a to this process is further established by administrating its mimic, which is sufficient to induce cardiac defects, and by using its antagomir to alleviate RBFox2 depletion-induced heart dysfunction. These findings elucidate a potential feed-forward mechanism to account for a critical transition to cardiac decompensation and suggest a potential therapeutic avenue against heart failure.
Keywords: EC coupling; Jph2; RBFox2; heart failure; miR-34a.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
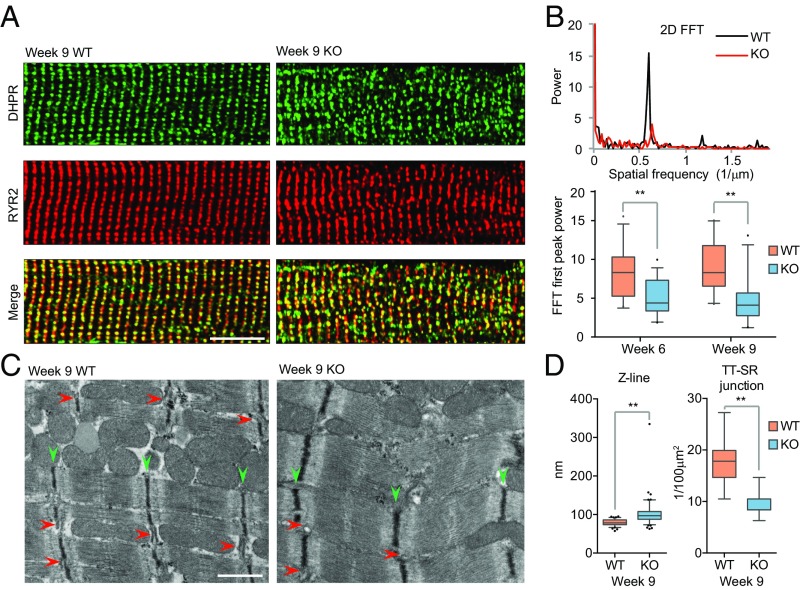
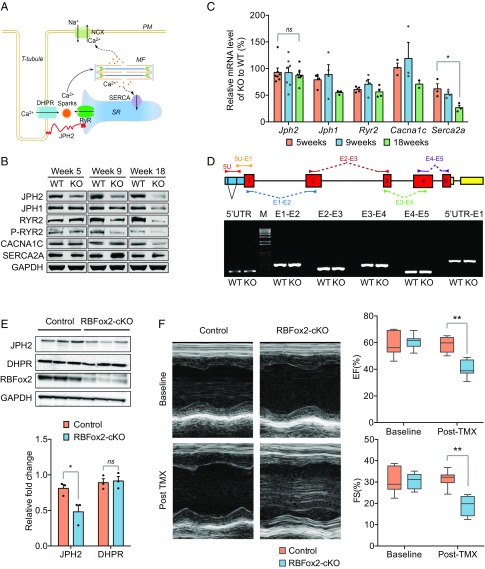
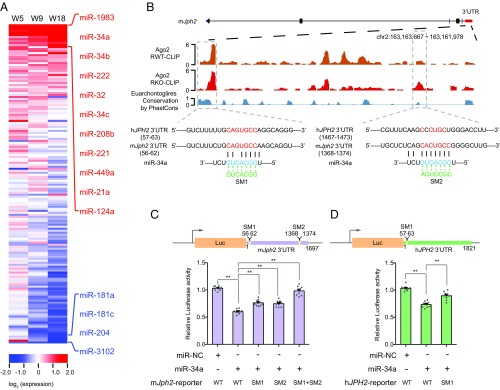
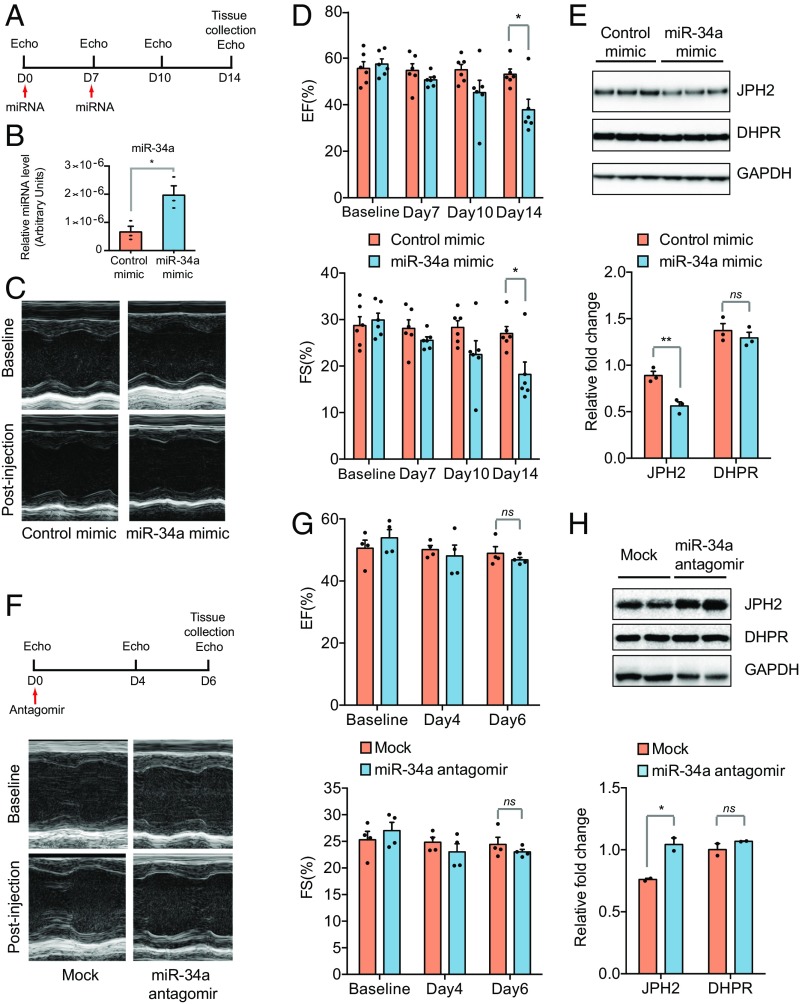
References
-
- Lakatta EG. Age-associated cardiovascular changes in health: Impact on cardiovascular disease in older persons. Heart Fail Rev. 2002;7:29–49. - PubMed
-
- Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: Cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
