

Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses
- PMID: 30867305
- PMCID: PMC6532088
- DOI: 10.1128/JVI.00354-19
Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses
Abstract
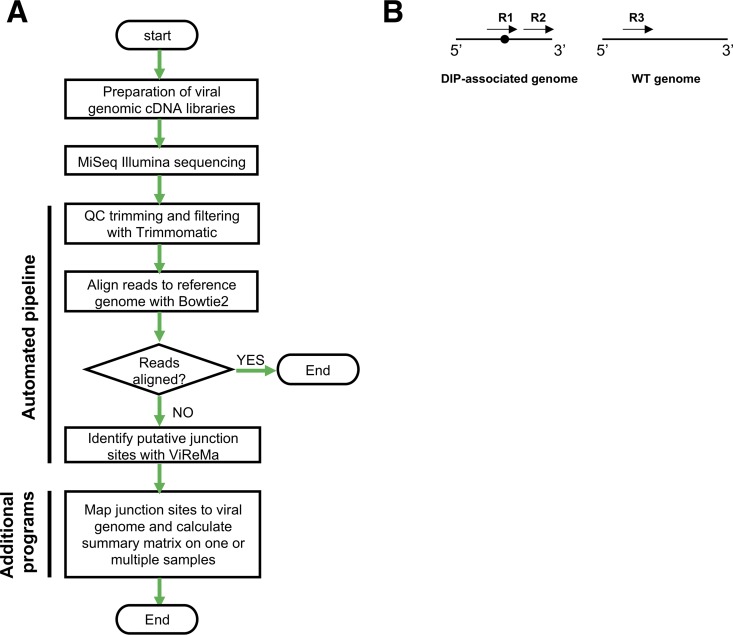
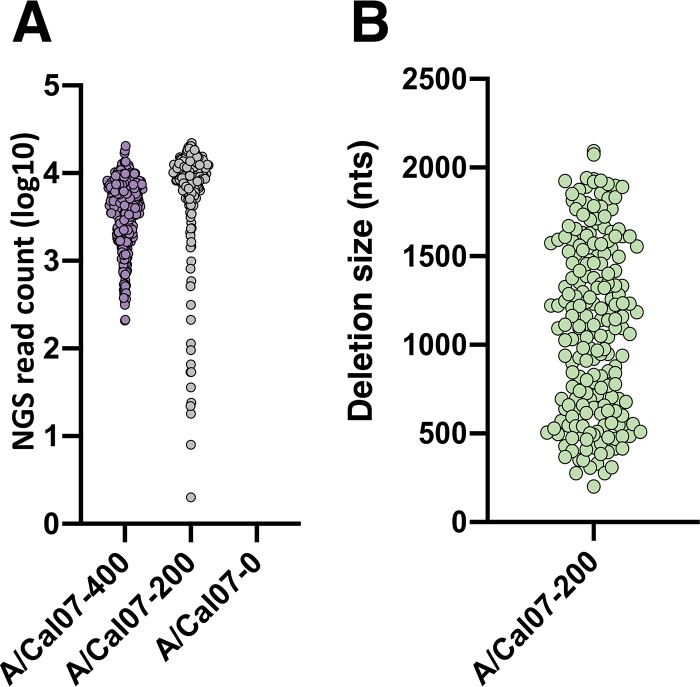
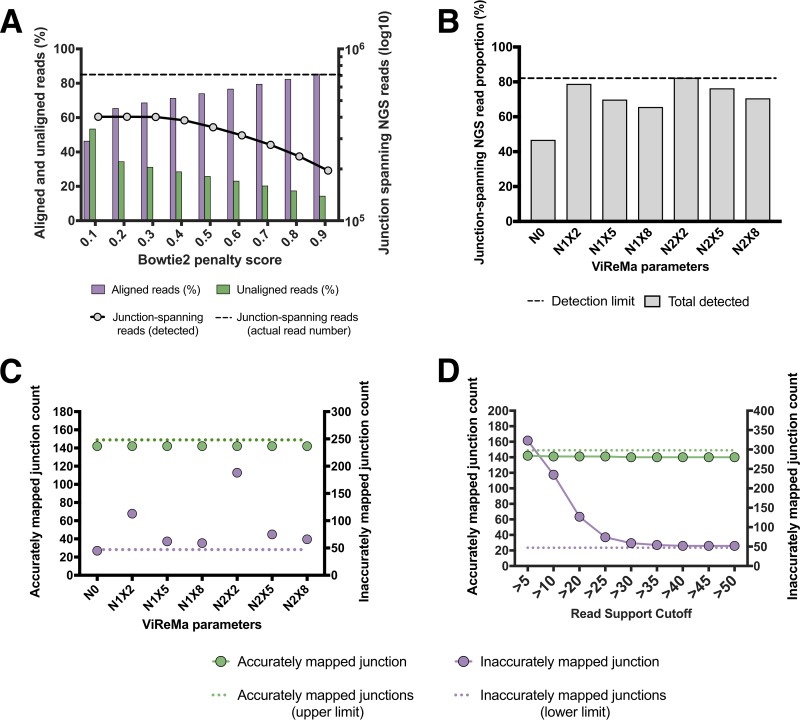
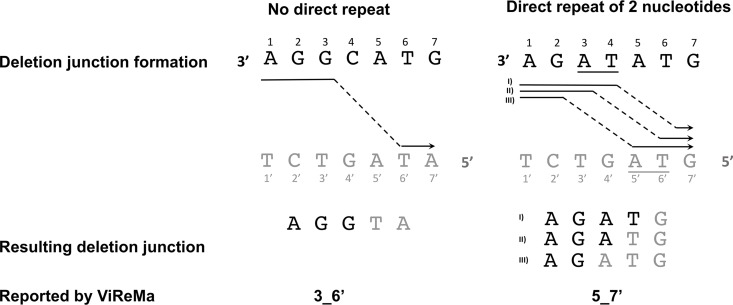
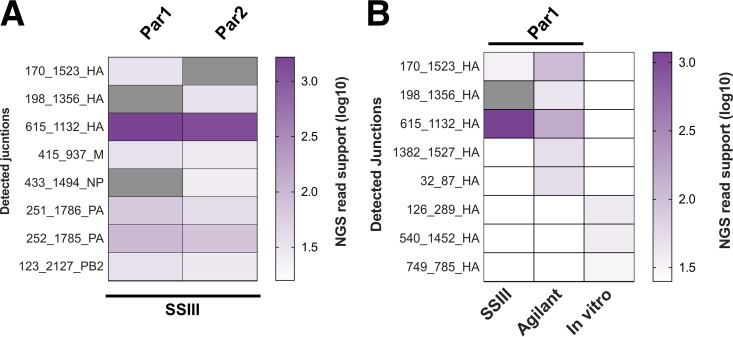
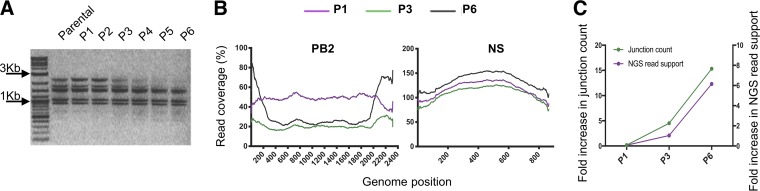
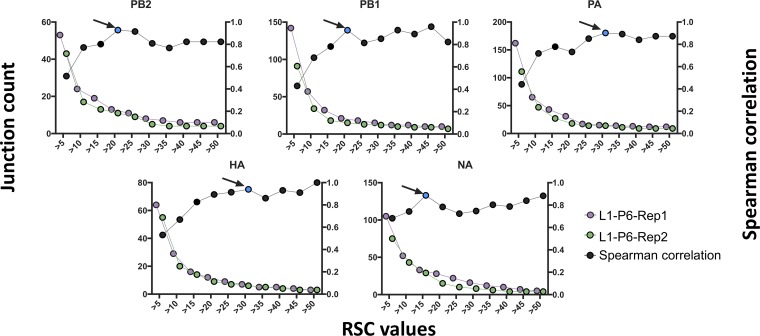
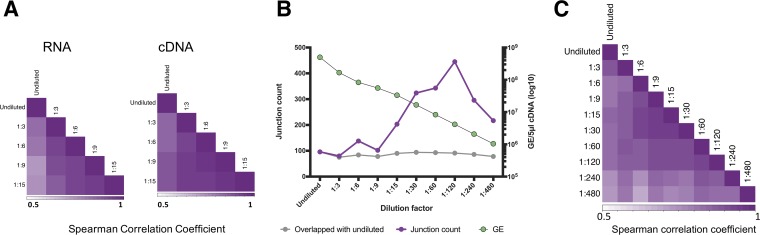
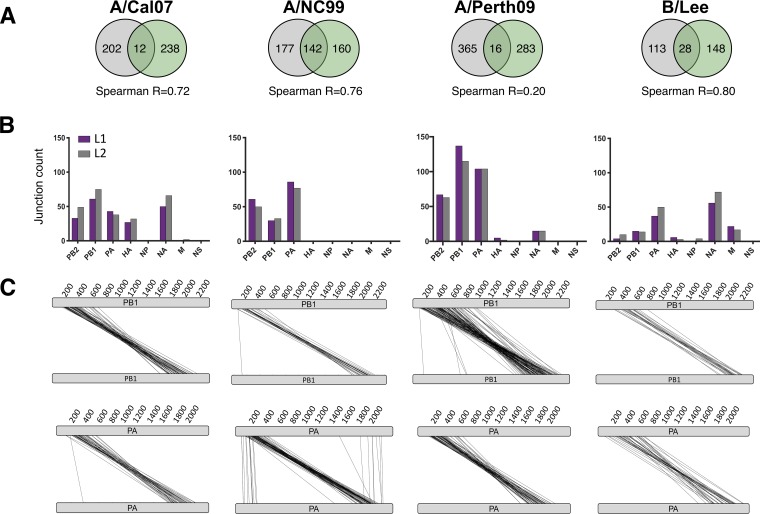
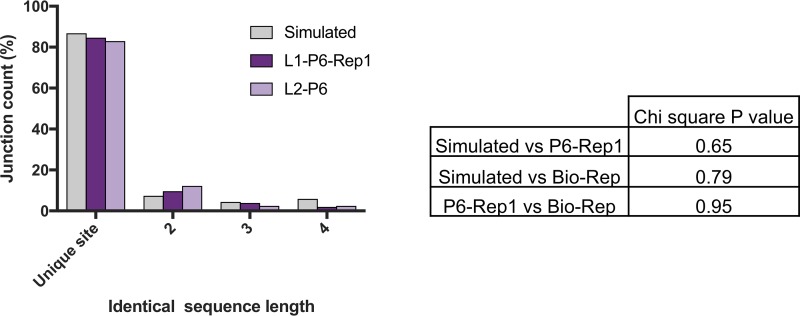
The mechanisms and consequences of defective interfering particle (DIP) formation during influenza virus infection remain poorly understood. The development of next-generation sequencing (NGS) technologies has made it possible to identify large numbers of DIP-associated sequences, providing a powerful tool to better understand their biological relevance. However, NGS approaches pose numerous technical challenges, including the precise identification and mapping of deletion junctions in the presence of frequent mutation and base-calling errors, and the potential for numerous experimental and computational artifacts. Here, we detail an Illumina-based sequencing framework and bioinformatics pipeline capable of generating highly accurate and reproducible profiles of DIP-associated junction sequences. We use a combination of simulated and experimental control data sets to optimize pipeline performance and demonstrate the absence of significant artifacts. Finally, we use this optimized pipeline to reveal how the patterns of DIP-associated junction formation differ between different strains and subtypes of influenza A and B viruses and to demonstrate how these data can provide insight into mechanisms of DIP formation. Overall, this work provides a detailed roadmap for high-resolution profiling and analysis of DIP-associated sequences within influenza virus populations.IMPORTANCE Influenza virus defective interfering particles (DIPs) that harbor internal deletions within their genomes occur naturally during infection in humans and during cell culture. They have been hypothesized to influence the pathogenicity of the virus; however, their specific function remains elusive. The accurate detection of DIP-associated deletion junctions is crucial for understanding DIP biology but is complicated by an array of technical issues that can bias or confound results. Here, we demonstrate a combined experimental and computational framework for detecting DIP-associated deletion junctions using next-generation sequencing (NGS). We detail how to validate pipeline performance and provide the bioinformatics pipeline for groups interested in using it. Using this optimized pipeline, we detect hundreds of distinct deletion junctions generated during infection with a diverse panel of influenza viruses and use these data to test a long-standing hypothesis concerning the molecular details of DIP formation.
Keywords: defective interfering particles; influenza; next-generation sequencing.
Copyright © 2019 American Society for Microbiology.
Figures
References
-
- von Magnus P. 1951. Propagation of the PR8 strain of influenza A virus in chick embryos. II. The formation of incomplete virus following inoculation of large doses of seed virus. Acta Pathol Microbiol Scand 28:278–293. - PubMed
-
- Nayak DP, Chambers TM, Akkina RK. 1985. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr Top Microbiol Immunol 114:103–151. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
