

Cellular DNA Topoisomerases Are Required for the Synthesis of Hepatitis B Virus Covalently Closed Circular DNA
- PMID: 30867306
- PMCID: PMC6532102
- DOI: 10.1128/JVI.02230-18
Cellular DNA Topoisomerases Are Required for the Synthesis of Hepatitis B Virus Covalently Closed Circular DNA
Abstract
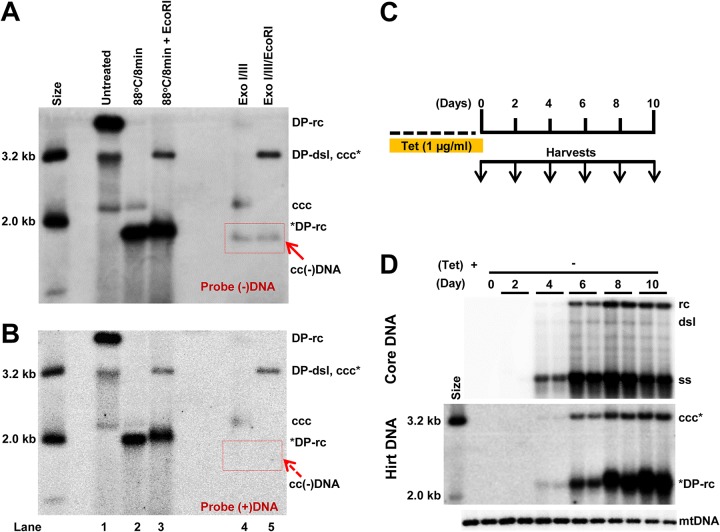
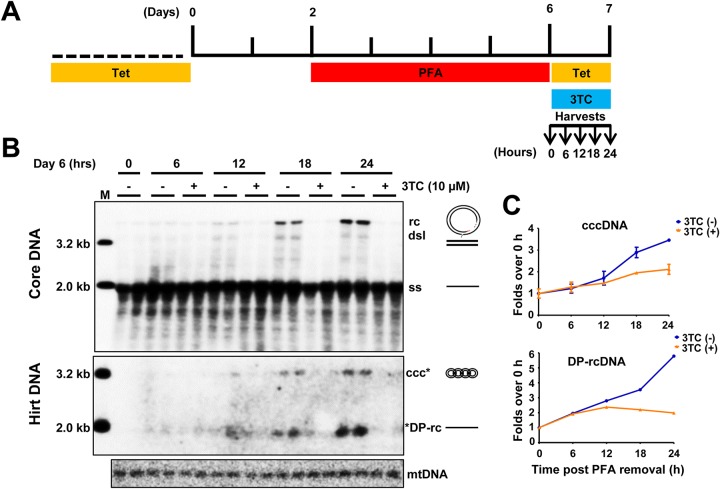
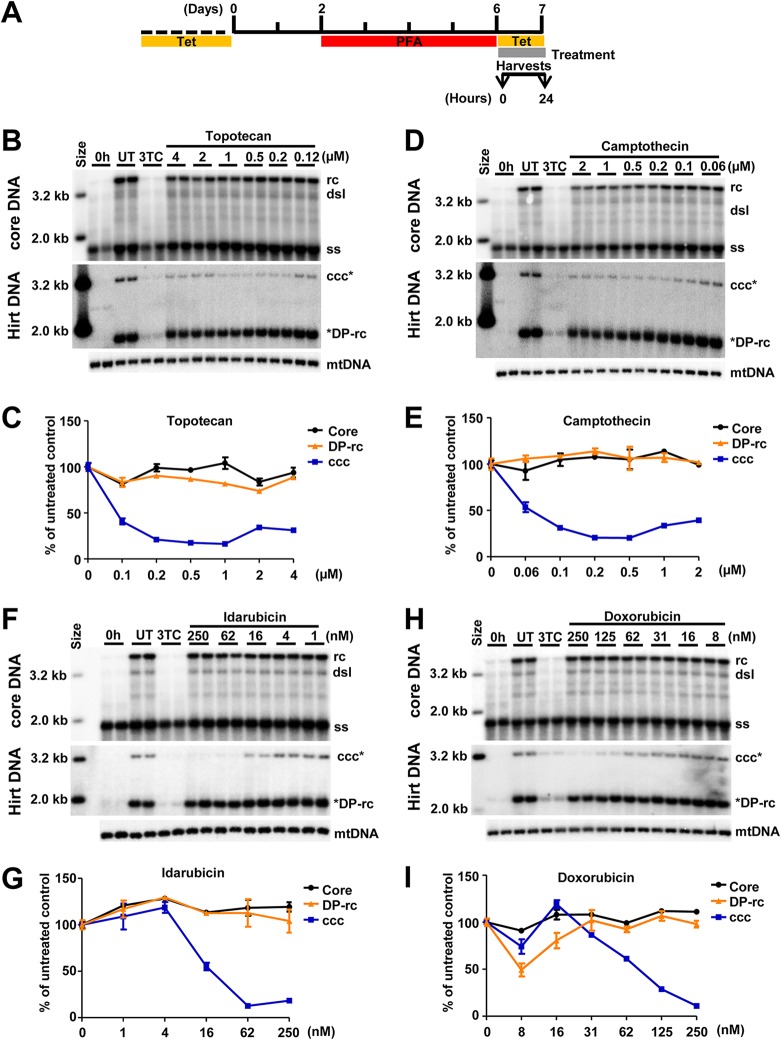
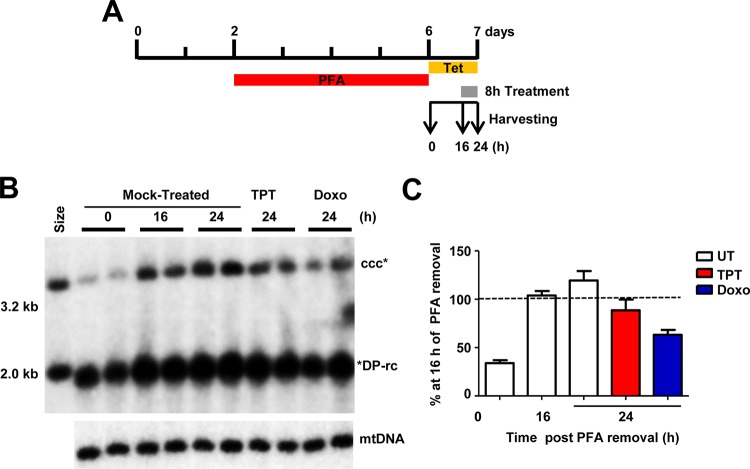
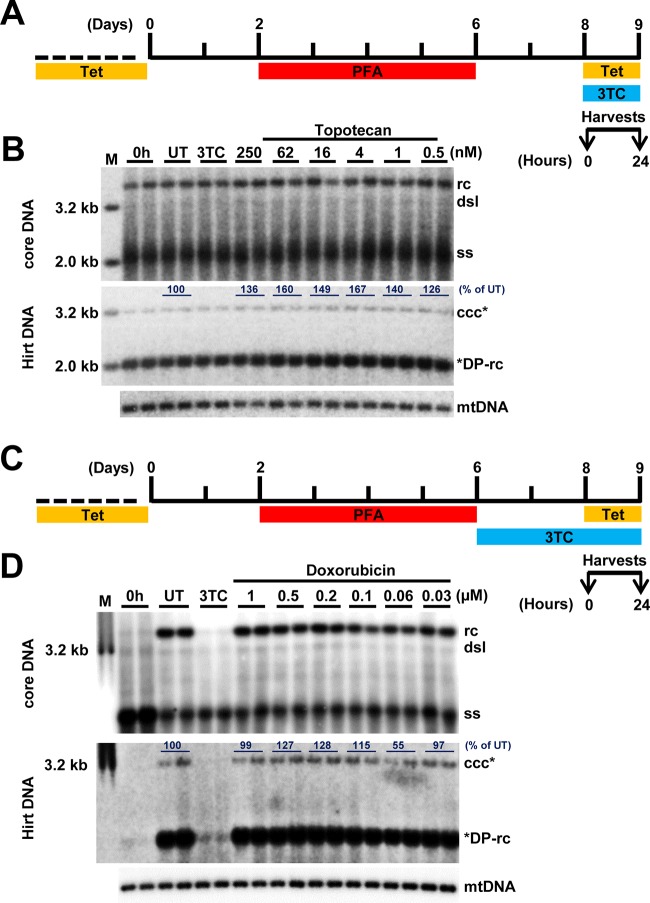
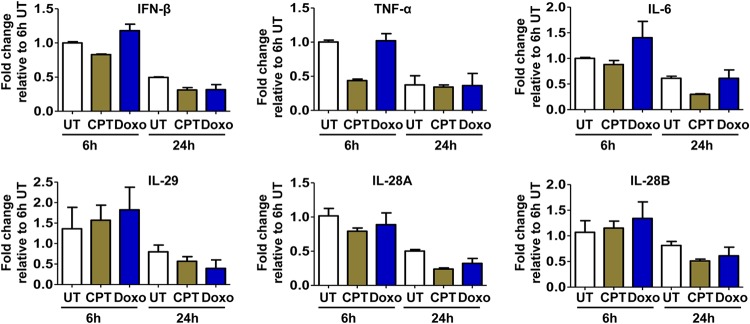
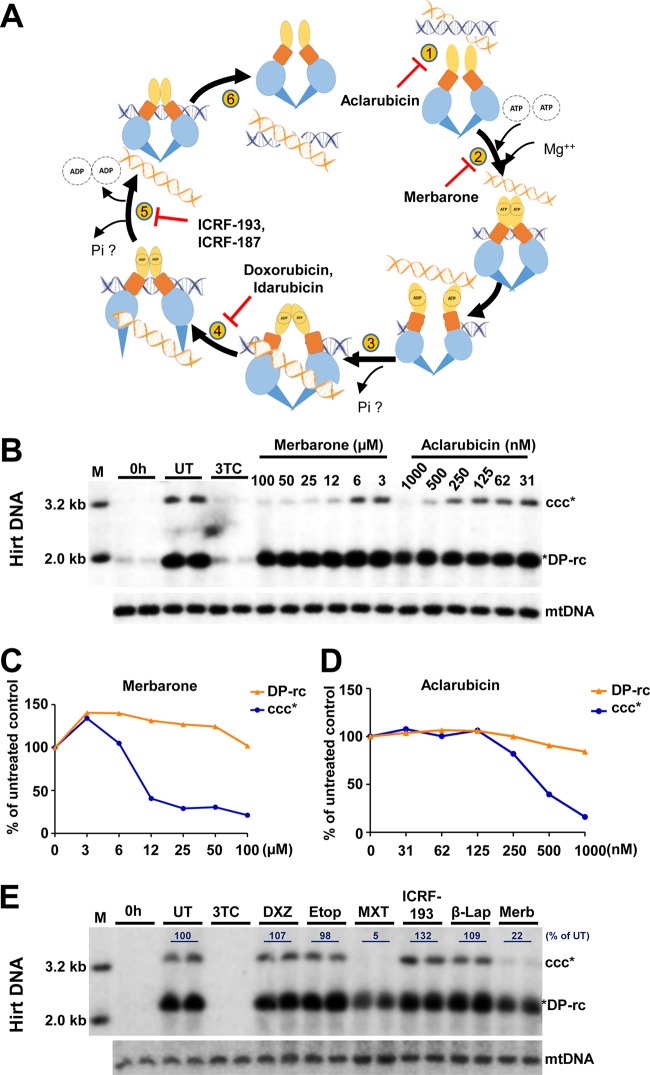
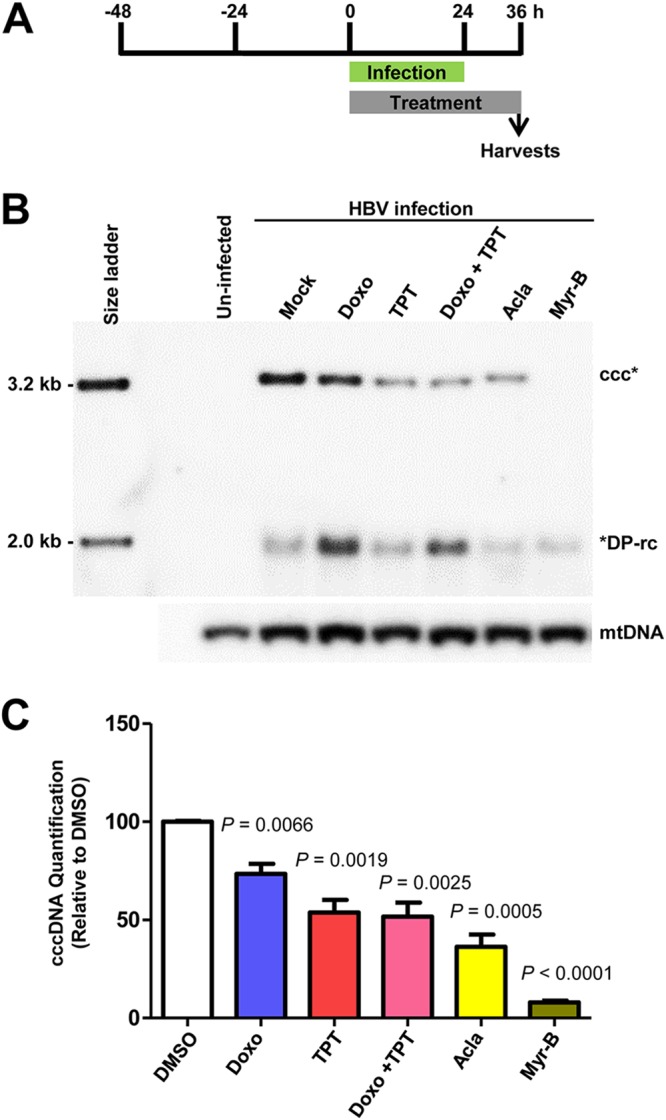
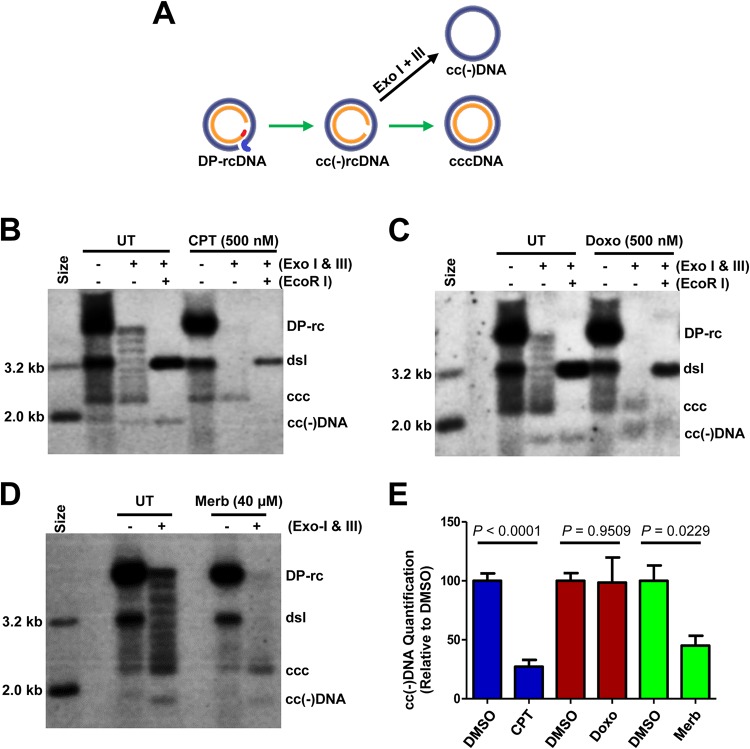
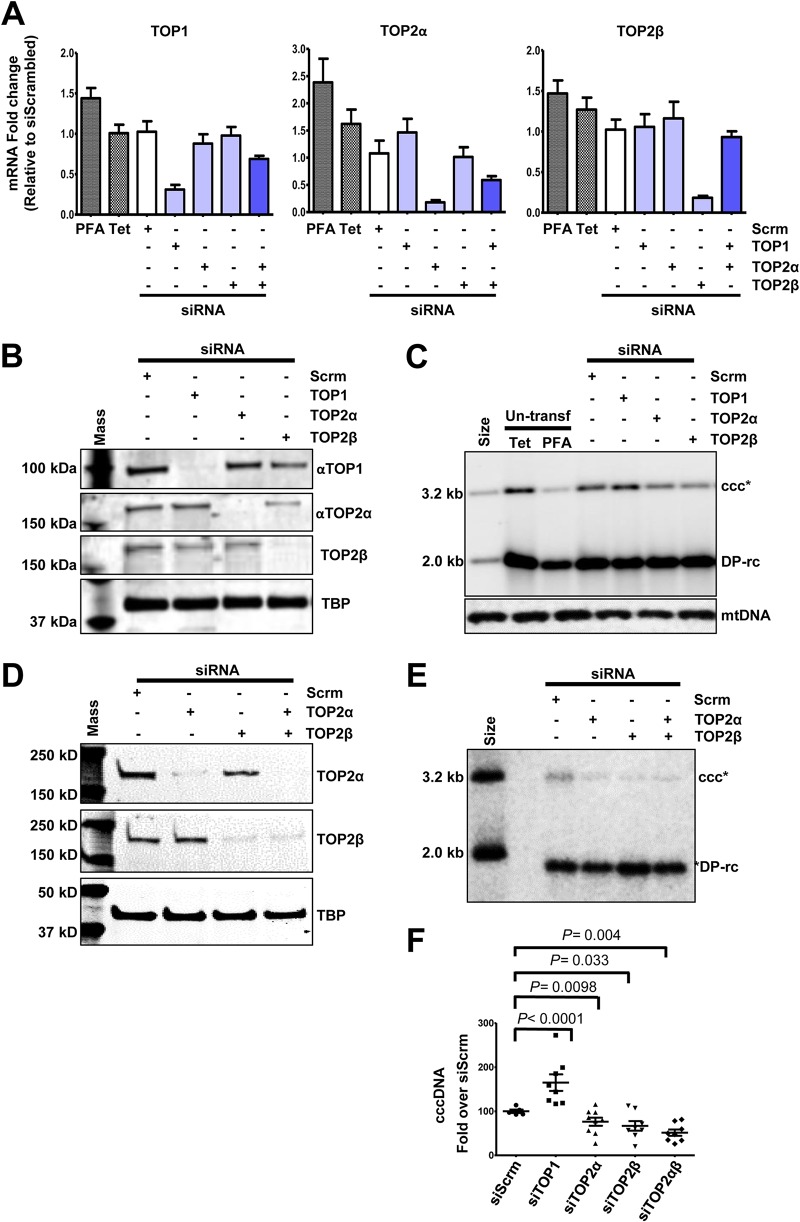
In order to identify host cellular DNA metabolic enzymes that are involved in the biosynthesis of hepatitis B virus (HBV) covalently closed circular (ccc) DNA, we developed a cell-based assay supporting synchronized and rapid cccDNA synthesis from intracellular progeny nucleocapsid DNA. This was achieved by arresting HBV DNA replication in HepAD38 cells with phosphonoformic acid (PFA), a reversible HBV DNA polymerase inhibitor, at the stage of single-stranded DNA and was followed by removal of PFA to allow the synchronized synthesis of relaxed circular DNA (rcDNA) and subsequent conversion into cccDNA within 12 to 24 h. This cccDNA formation assay allows systematic screening of the effects of small molecular inhibitors of DNA metabolic enzymes on cccDNA synthesis but avoids cytotoxic effects upon long-term treatment. Using this assay, we found that all the tested topoisomerase I and II (TOP1 and TOP2, respectively) poisons as well as topoisomerase II DNA binding and ATPase inhibitors significantly reduced the levels of cccDNA. It was further demonstrated that these inhibitors also disrupted cccDNA synthesis during de novo HBV infection of HepG2 cells expressing sodium taurocholate cotransporting polypeptide (NTCP). Mechanistic analyses indicate that whereas TOP1 inhibitor treatment prevented the production of covalently closed negative-strand rcDNA, TOP2 inhibitors reduced the production of this cccDNA synthesis intermediate to a lesser extent. Moreover, small interfering RNA (siRNA) knockdown of topoisomerase II significantly reduced cccDNA amplification. Taking these observations together, our study demonstrates that topoisomerase I and II may catalyze distinct steps of HBV cccDNA synthesis and that pharmacologic targeting of these cellular enzymes may facilitate the cure of chronic hepatitis B.IMPORTANCE Persistent HBV infection relies on stable maintenance and proper functioning of a nuclear episomal form of the viral genome called cccDNA, the most stable HBV replication intermediate. One of the major reasons for the failure of currently available antiviral therapeutics to cure chronic HBV infection is their inability to eradicate or inactivate cccDNA. We report here a chemical genetics approach to identify host cellular factors essential for the biosynthesis and maintenance of cccDNA and reveal that cellular DNA topoisomerases are required for both de novo synthesis and intracellular amplification of cccDNA. This approach is suitable for systematic screening of compounds targeting cellular DNA metabolic enzymes and chromatin remodelers for their ability to disrupt cccDNA biosynthesis and function. Identification of key host factors required for cccDNA metabolism and function will reveal molecular targets for developing curative therapeutics of chronic HBV infection.
Keywords: DNA topoisomerase; antiviral agents; cccDNA; hepatitis B virus.
Copyright © 2019 American Society for Microbiology.
Figures
References
-
- GBD 2013 Mortality and Causes of Death Collaborators. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385:117–171. doi: 10.1016/S0140-6736(14)61682-2. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
