New insights from uncultivated genomes of the global human gut microbiome
- PMID: 30867587
- PMCID: PMC6784871
- DOI: 10.1038/s41586-019-1058-x
New insights from uncultivated genomes of the global human gut microbiome
Abstract
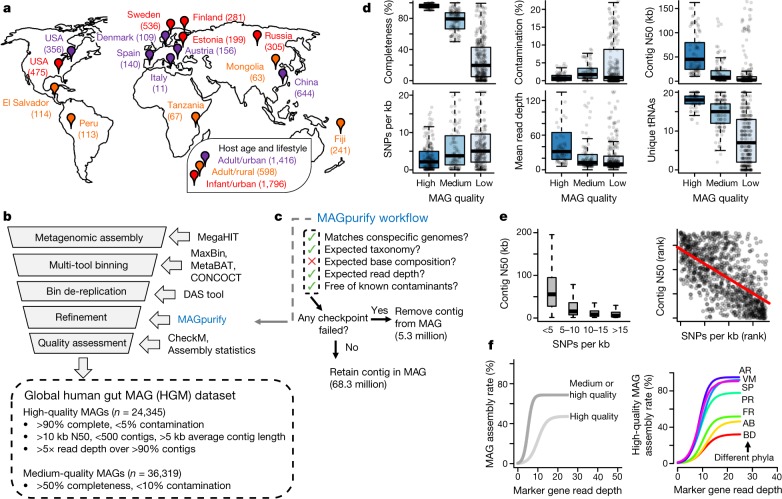
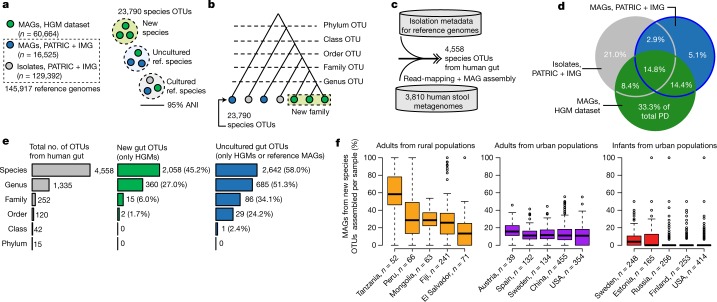
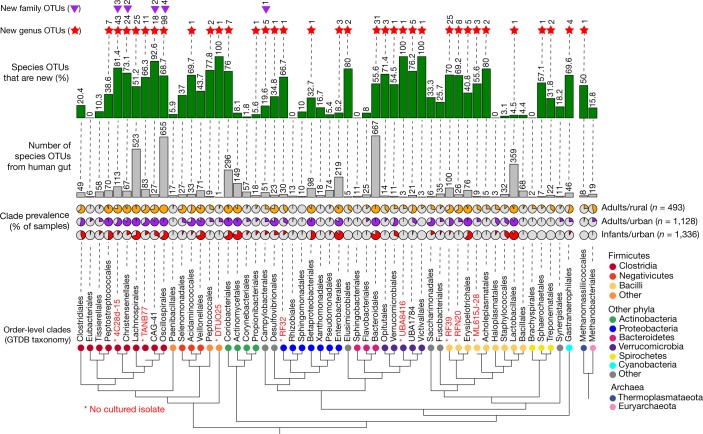
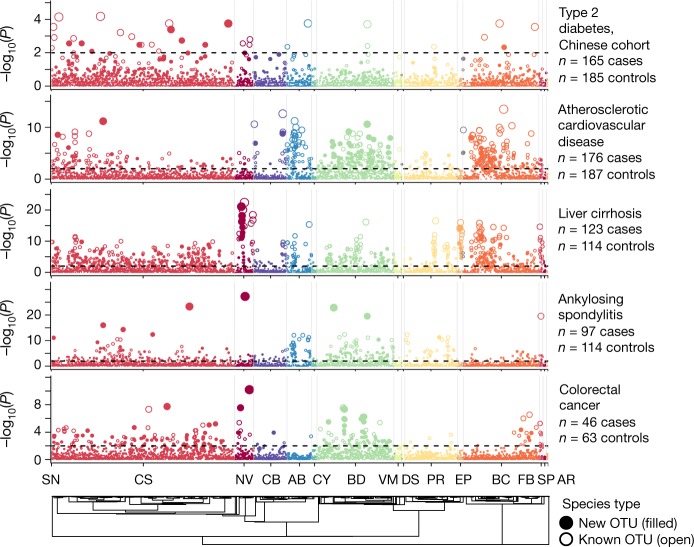
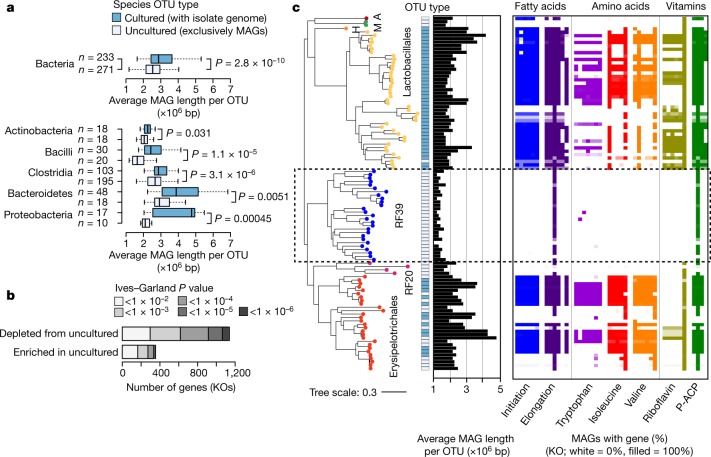
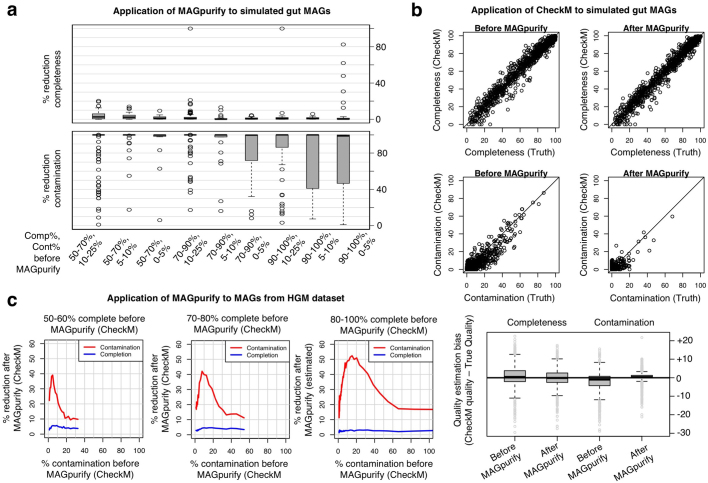
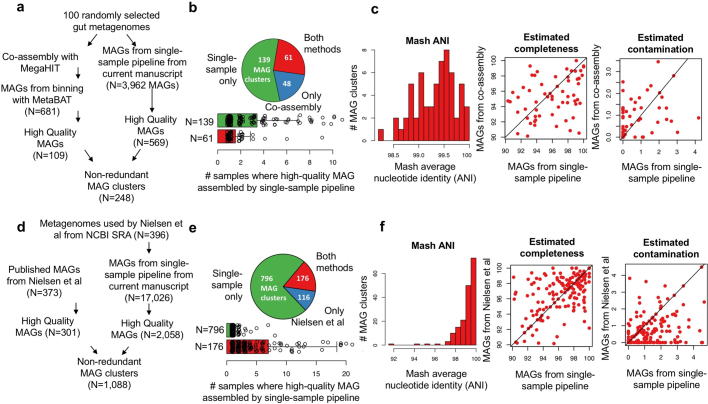
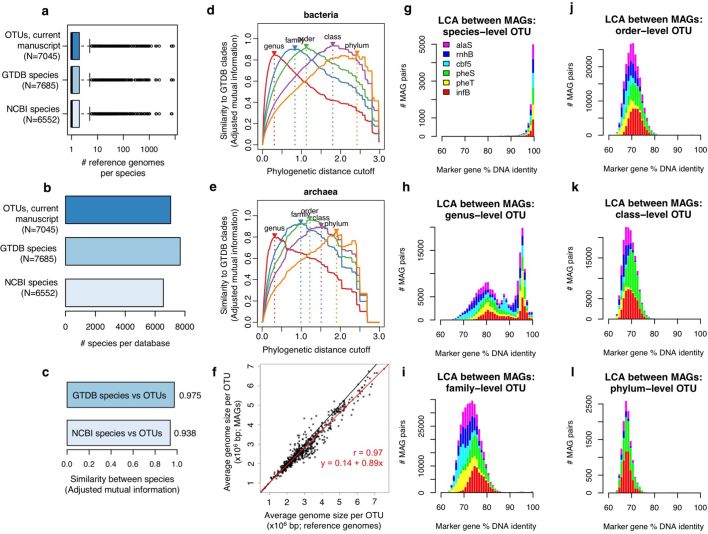
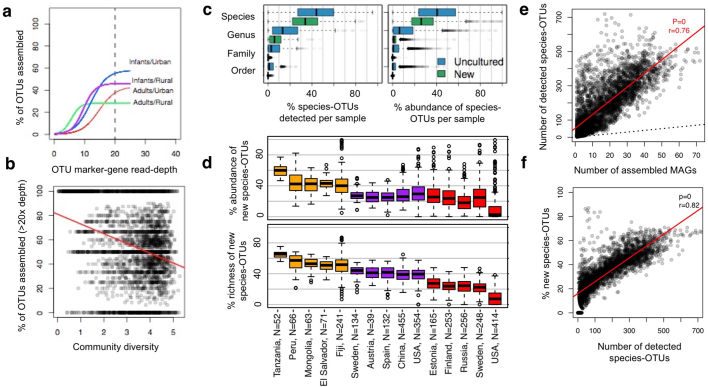
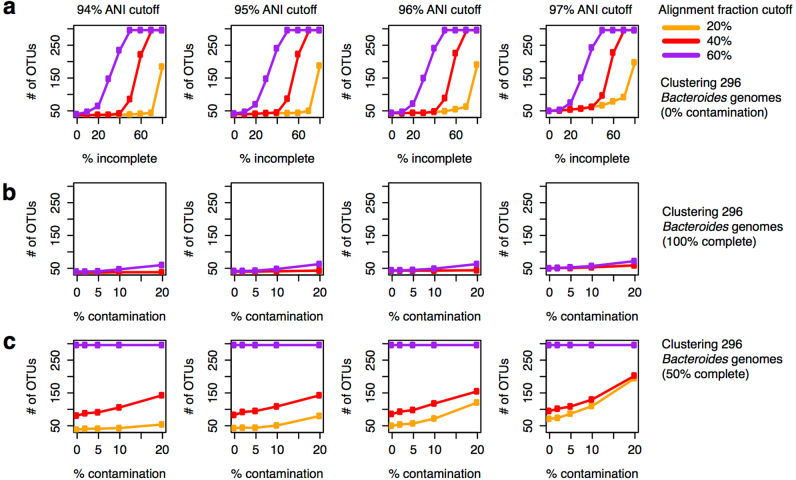
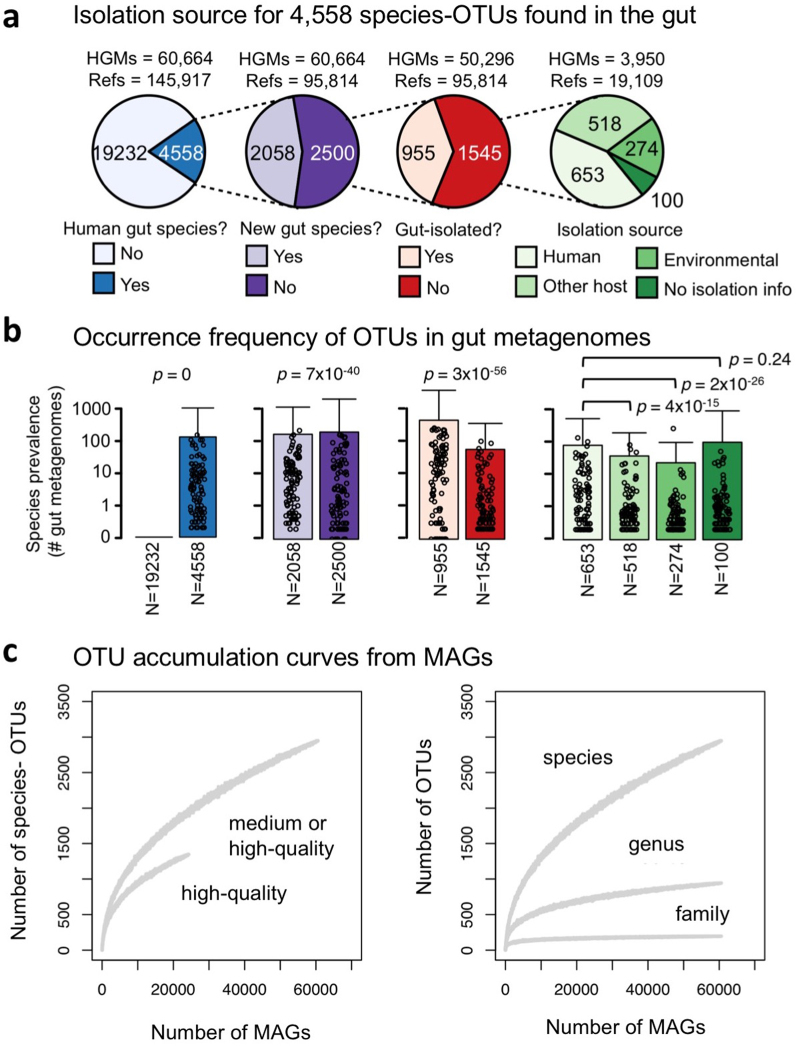
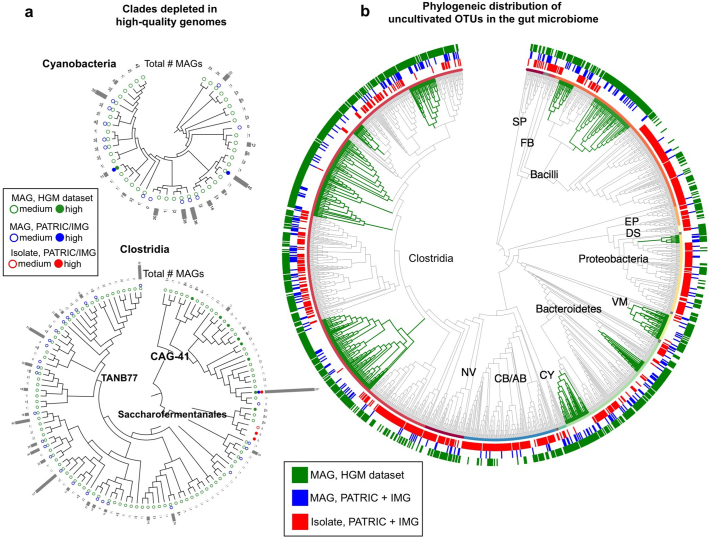
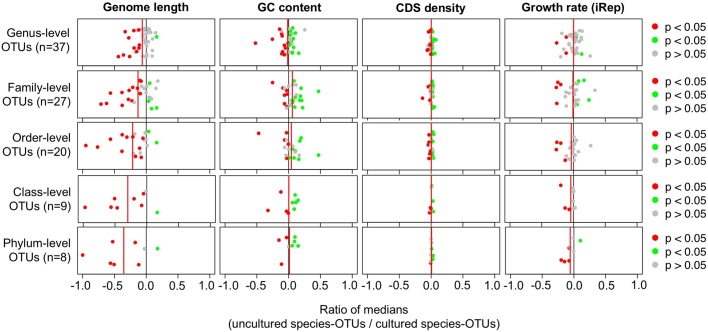
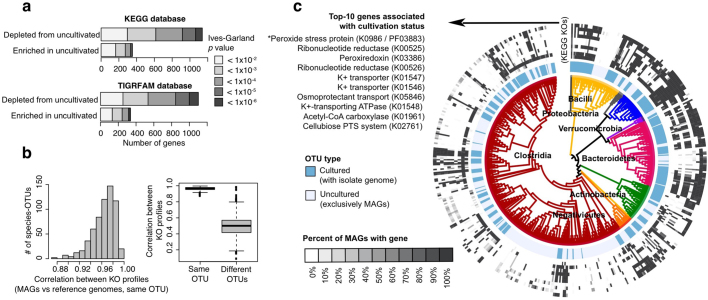
The genome sequences of many species of the human gut microbiome remain unknown, largely owing to challenges in cultivating microorganisms under laboratory conditions. Here we address this problem by reconstructing 60,664 draft prokaryotic genomes from 3,810 faecal metagenomes, from geographically and phenotypically diverse humans. These genomes provide reference points for 2,058 newly identified species-level operational taxonomic units (OTUs), which represents a 50% increase over the previously known phylogenetic diversity of sequenced gut bacteria. On average, the newly identified OTUs comprise 33% of richness and 28% of species abundance per individual, and are enriched in humans from rural populations. A meta-analysis of clinical gut-microbiome studies pinpointed numerous disease associations for the newly identified OTUs, which have the potential to improve predictive models. Finally, our analysis revealed that uncultured gut species have undergone genome reduction that has resulted in the loss of certain biosynthetic pathways, which may offer clues for improving cultivation strategies in the future.
Conflict of interest statement
K.S.P. is on the advisory boards of uBiome and Phylagen.
Figures