

The natural history of progressive fibrosing interstitial lung diseases
- PMID: 30871560
- PMCID: PMC6417262
- DOI: 10.1186/s12931-019-1022-1
The natural history of progressive fibrosing interstitial lung diseases
Abstract
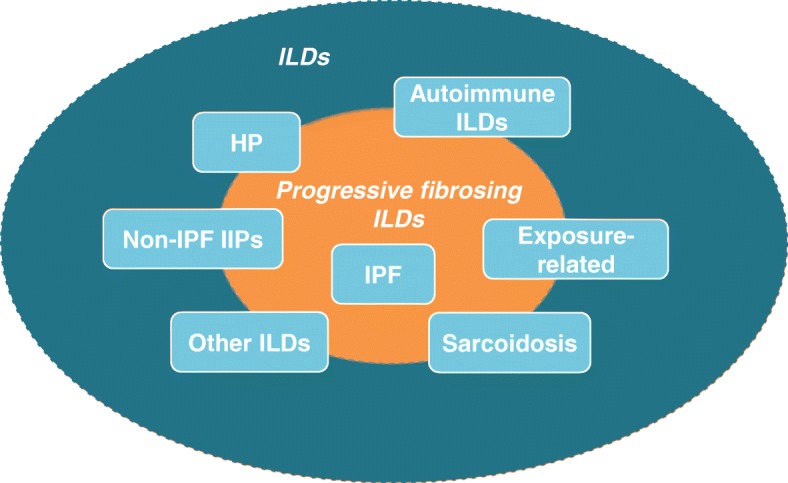
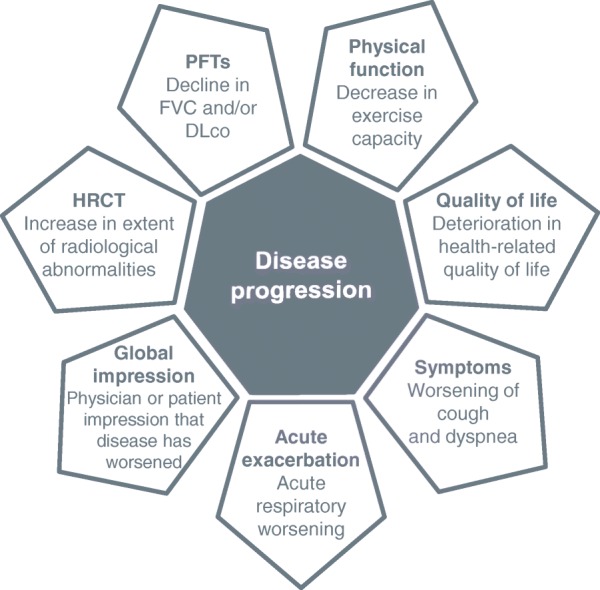
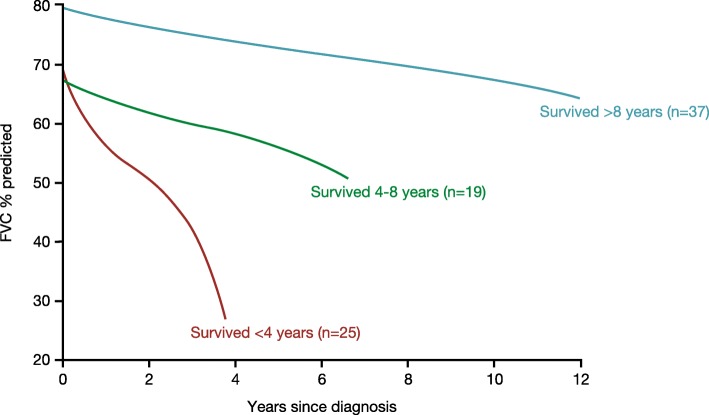
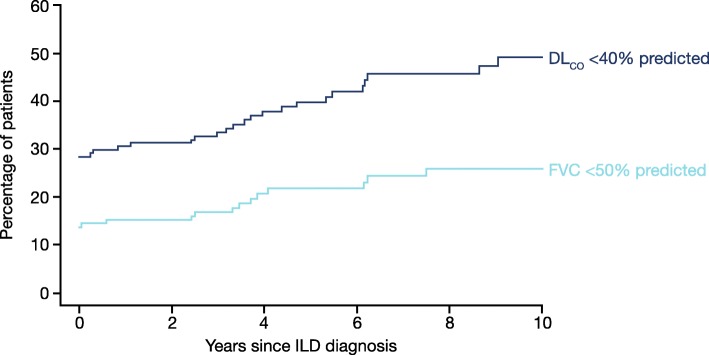
A proportion of patients with certain types of interstitial lung disease (ILD), including chronic hypersensitivity pneumonitis and ILDs associated with autoimmune diseases, develop a progressive fibrosing phenotype that shows similarities in clinical course to idiopathic pulmonary fibrosis. Irrespective of the clinical diagnosis, these progressive fibrosing ILDs show commonalities in the underlying pathogenetic mechanisms that drive a self-sustaining process of pulmonary fibrosis. The natural history of progressive fibrosing ILDs is characterized by decline in lung function, worsening of symptoms and health-related quality of life, and early mortality. Greater impairment in forced vital capacity or diffusion capacity of the lungs for carbon monoxide, and a greater extent of fibrotic changes on a computed tomography scan, are predictors of mortality in patients with fibrosing ILDs. However, the course of these diseases is heterogenous and cannot accurately be predicted for an individual patient. Data from ongoing clinical trials and patient registries will provide a better understanding of the clinical course and impact of progressive fibrosing ILDs.
Keywords: Connective tissue diseases; Mortality; Pulmonary fibrosis; Rheumatic diseases; Systemic sclerosis; Vital capacity.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Martin Kolb reports receipt of grants and personal fees from Boehringer Ingelheim and Roche; personal fees from GlaxoSmithKline, Gilead, AstraZeneca, ProMetic and Genoa; and grants from Actelion, Respivert, the Canadian Institutes of Health Research (MOP-136950), and the Canadian Pulmonary Fibrosis Foundation. Martina Vašáková has received payment for consultancy, lectures and advisory board attendance from Roche and Boehringer Ingelheim and has received research grants from Roche.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. - PubMed
-
- Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ; IPF Consensus Working Group. What's in a name? That which we call IPF, by any other name would act the same. Eur Respir J 2018;51(5) pii: 1800692. - PubMed
-
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. - PubMed
-
- Winstone TA, Assayag D, Wilcox PG, Dunne JV, Hague CJ, Leipsic J, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 2014;146(2):422–436. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
