

Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes
- PMID: 30872533
- PMCID: PMC6532986
- DOI: 10.1126/science.aau1330
Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes
Abstract
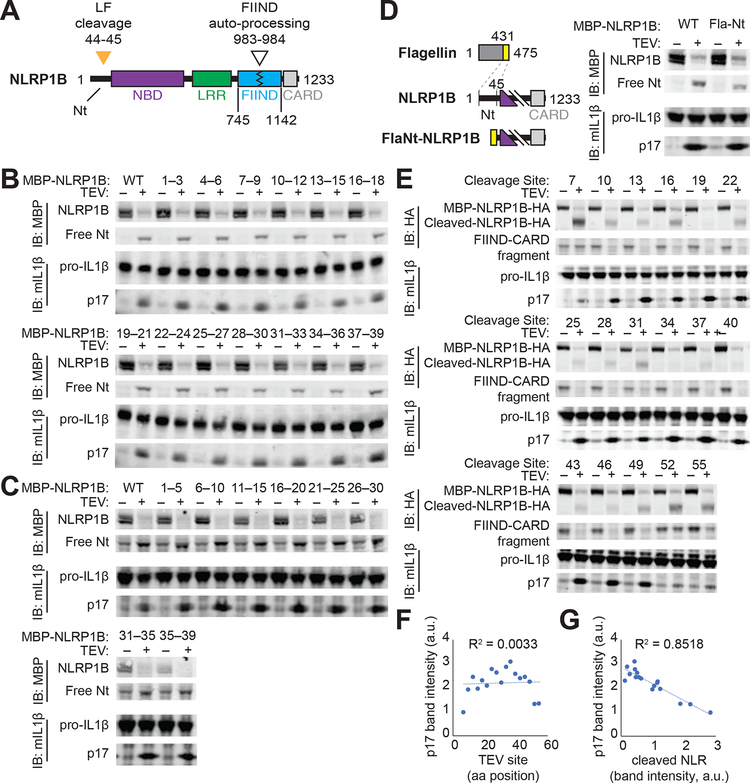
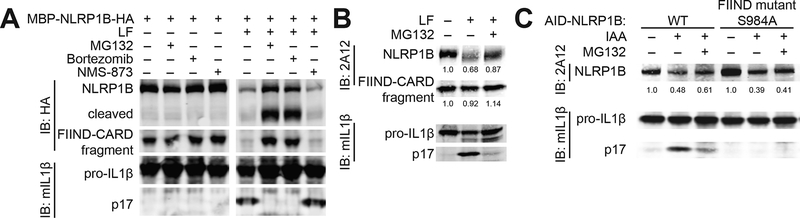
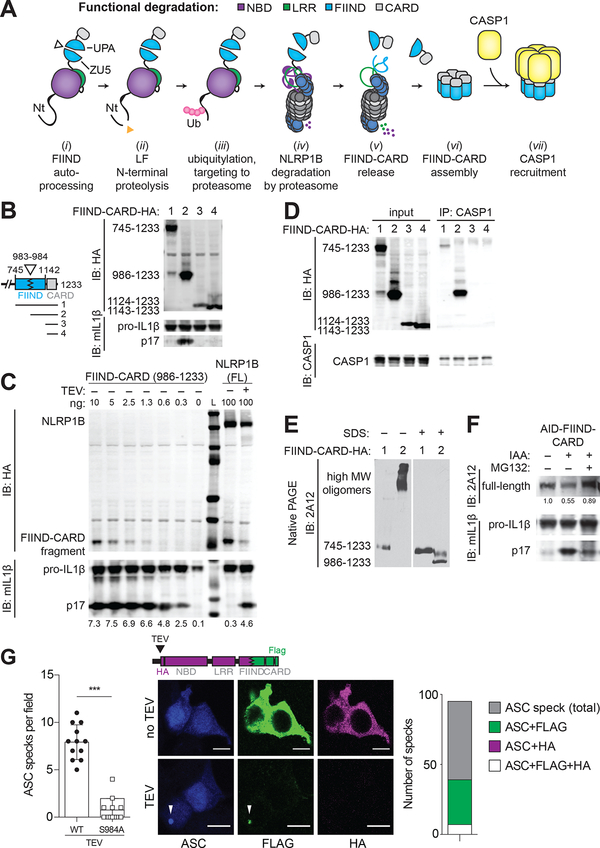
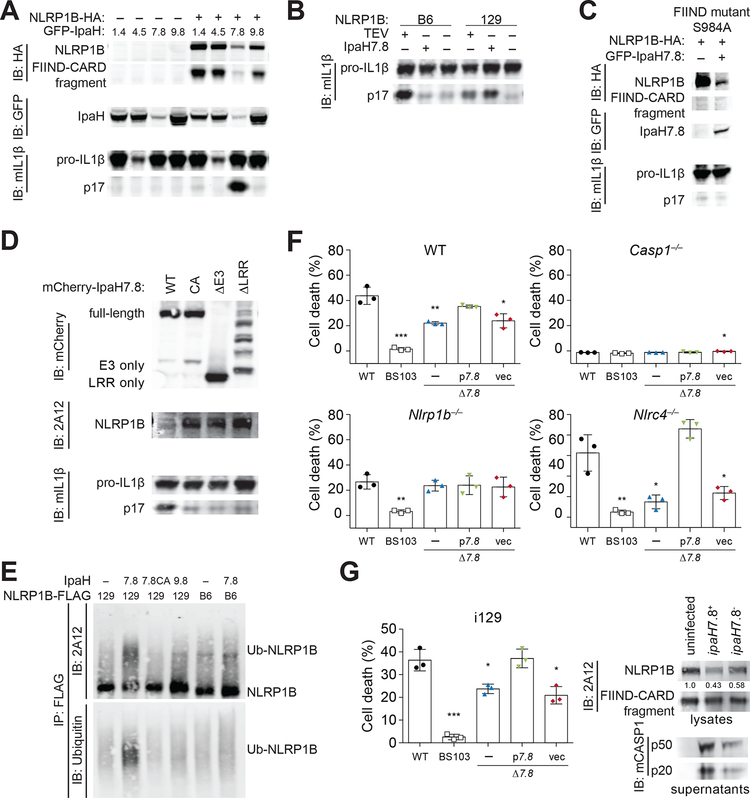
Inflammasomes are multiprotein platforms that initiate innate immunity by recruitment and activation of caspase-1. The NLRP1B inflammasome is activated upon direct cleavage by the anthrax lethal toxin protease. However, the mechanism by which cleavage results in NLRP1B activation is unknown. In this study, we find that cleavage results in proteasome-mediated degradation of the amino-terminal domains of NLRP1B, liberating a carboxyl-terminal fragment that is a potent caspase-1 activator. Proteasome-mediated degradation of NLRP1B is both necessary and sufficient for NLRP1B activation. Consistent with our functional degradation model, we identify IpaH7.8, a Shigella flexneri ubiquitin ligase secreted effector, as an enzyme that induces NLRP1B degradation and activation. Our results provide a unified mechanism for NLRP1B activation by diverse pathogen-encoded enzymatic activities.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
Functional degradation ignites the inflammasome.Nat Rev Immunol. 2019 Jun;19(6):349. doi: 10.1038/s41577-019-0169-9. Nat Rev Immunol. 2019. PMID: 31000778 No abstract available.
-
Cell Death: N-degrons Fine-Tune Pyroptotic Cell Demise.Curr Biol. 2019 Jun 17;29(12):R588-R591. doi: 10.1016/j.cub.2019.05.004. Curr Biol. 2019. PMID: 31211982
References
-
- Janeway CA Jr., Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54 Pt 1, 1–13 (1989). - PubMed
-
- Jones JD, Dangl JL, The plant immune system. Nature 444, 323–329 (2006). - PubMed
-
- Blander JM, Sander LE, Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat. Rev. Immunol 12, 215–225 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
