

Ultra-fast and automated immunohistofluorescent multistaining using a microfluidic tissue processor
- PMID: 30872751
- PMCID: PMC6418167
- DOI: 10.1038/s41598-019-41119-y
Ultra-fast and automated immunohistofluorescent multistaining using a microfluidic tissue processor
Abstract
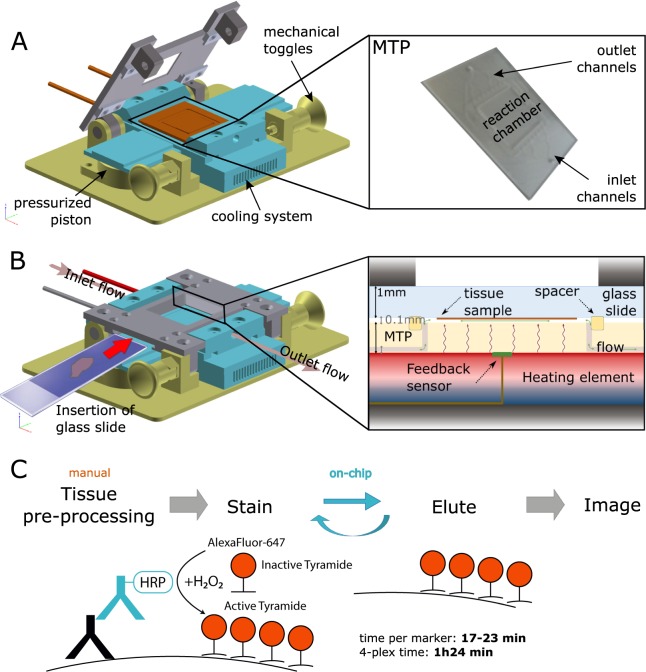
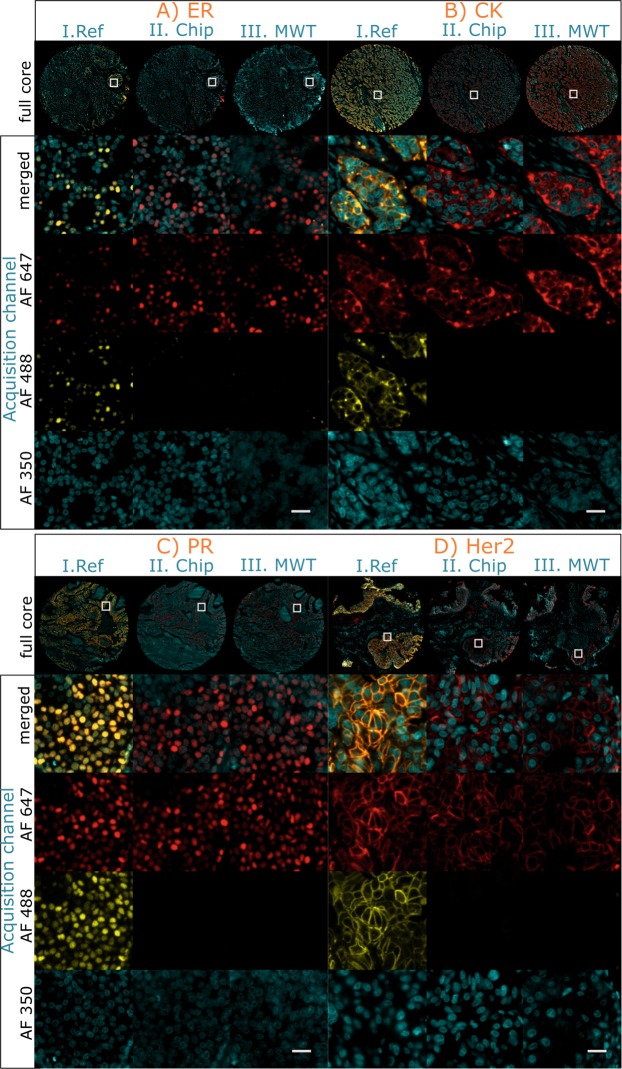
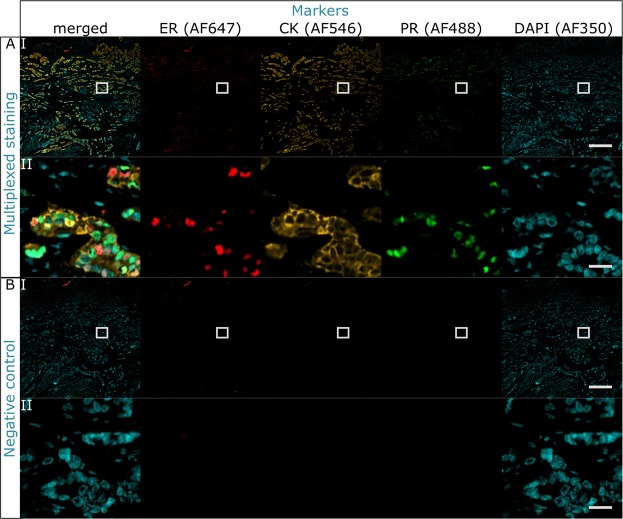
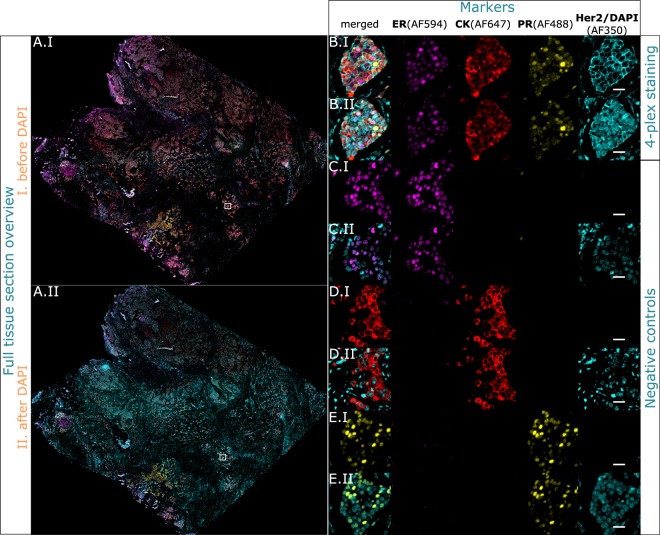
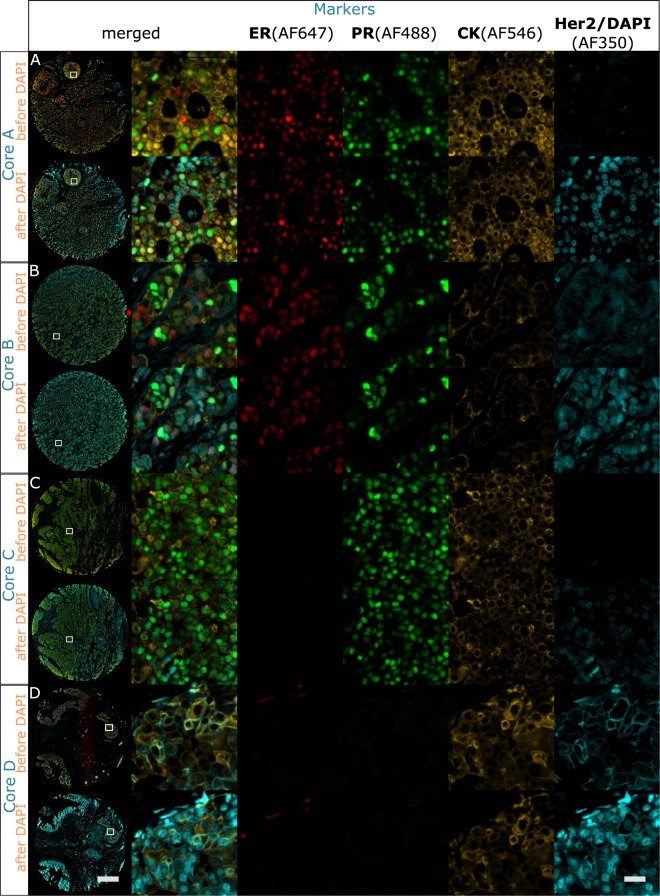
Multistaining of a tissue section targeting multiple markers allows to reveal complex interplays in a tumor environment. However, the resource-intensive and impractically long nature of iterative multiplexed immunostainings prohibits its practical implementation in daily routine, even when using work-flow automation systems. Here, we report a fully automated and ultra-fast multistaining using a microfluidic tissue processor (MTP) in as short as 20 minutes per marker, by immunofluorescent staining employing commercially available tyramide signal amplification polymer precipitation by horse-radish peroxidase (HRP) activation. The reported duration includes (i) 15 minutes for the entire fluidic exchange and reagent incubation necessary for the immunostaining and (ii) 5 minutes for the heat-induced removal of the applied antibodies. Using the automated MTP, we demonstrated a 4-plex automated multistaining with clinically relevant biomarkers within 84 minutes, showing perfect agreement with the state-of-the-art microwave treatment antibody removal. The presented HRP-based method is in principle extendable to multistaining by both tyramides accommodating higher number of fluorescent channels and multi-color chromogenic staining. We anticipate that our automated multi-staining with a turn-around time shorter than existing monoplex immunohistochemistry methods has the potential to enable multistaining in routine without disturbing the current laboratory workflow, opening perspectives for implementation of -omics approaches in tissue diagnostics.
Conflict of interest statement
All the authors are employed by Lunaphore Technologies SA, which filed a patent application covering a portion of the subject matter of this manuscript and which is commercializing the MTP-based technology. D.G.D. and A.T.C. have equity interest in Lunaphore Technologies SA. G.C. has stock options in Lunaphore Technologies SA.
Figures
References
-
- Jürgensmeier, J. M., Eder, J. P. & Herbst, R. S. New Strategies in Personalized Medicine for Solid Tumors: Molecular Markers and Clinical Trial Designs. Clinical cancer research: an official journal of the American Association for Cancer Research20, 4425–4435, http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5369358/, 10.1158/1078-0432.CCR-13-0753 (2014). - PMC - PubMed
-
- Hicks DG. T. P. Molecular classifications of breast carcinoma: is immunohistochemistry a viable alternative to other molecular methodologies? Connection. 2009;13:31–34.
-
- Tan, D. & Zander, D. S. Immunohistochemistry for Assessment of Pulmonary and Pleural Neoplasms: A Review and Update. International Journal of Clinical and Experimental Pathology1, 19–31, http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2480532/ (2008). - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
