

The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism
- PMID: 30873120
- PMCID: PMC6401612
- DOI: 10.3389/fendo.2019.00111
The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism
Abstract
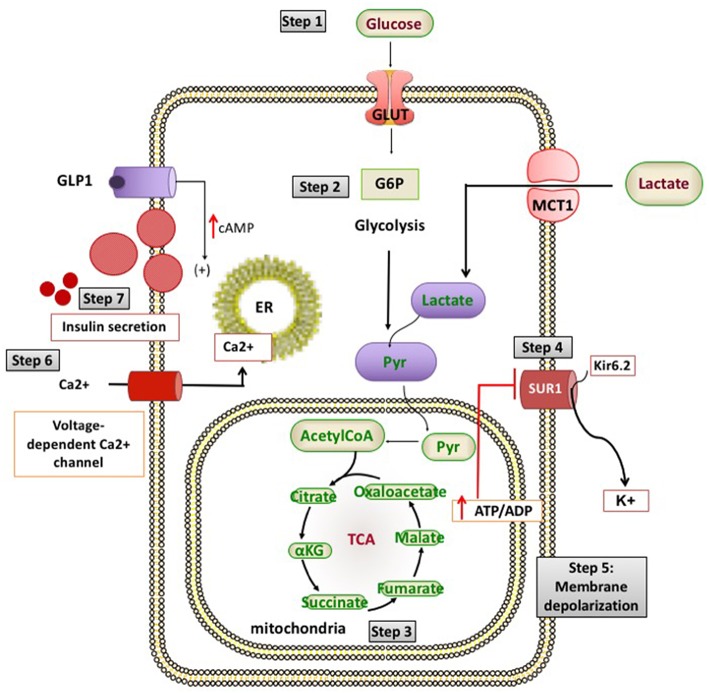
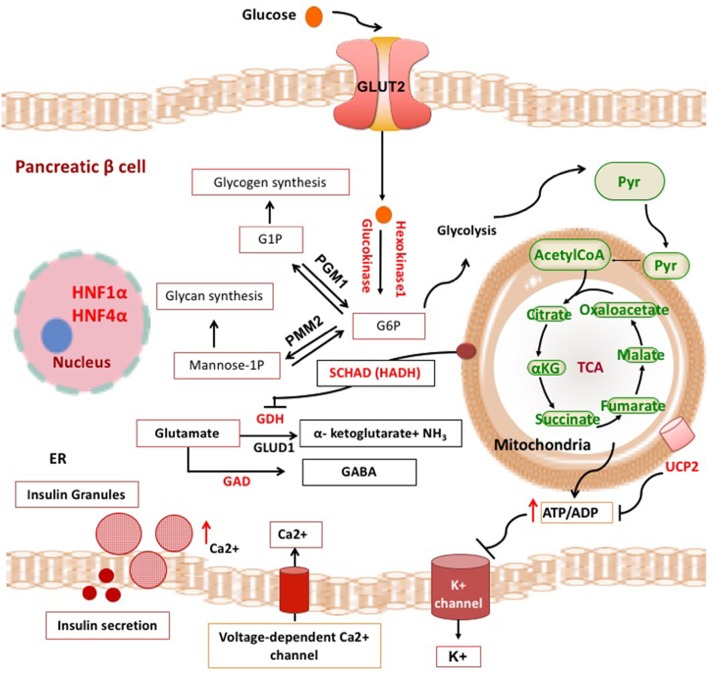
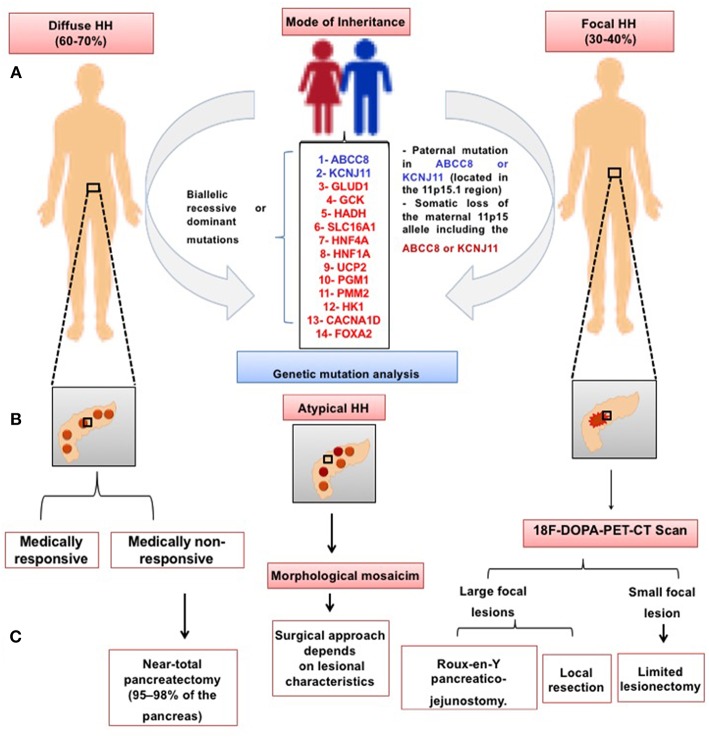
Congenital hyperinsulinism (CHI) is a heterogenous and complex disorder in which the unregulated insulin secretion from pancreatic beta-cells leads to hyperinsulinaemic hypoglycaemia. The severity of hypoglycaemia varies depending on the underlying molecular mechanism and genetic defects. The genetic and molecular causes of CHI include defects in pivotal pathways regulating the secretion of insulin from the beta-cell. Broadly these genetic defects leading to unregulated insulin secretion can be grouped into four main categories. The first group consists of defects in the pancreatic KATP channel genes (ABCC8 and KCNJ11). The second and third categories of conditions are enzymatic defects (such as GDH, GCK, HADH) and defects in transcription factors (for example HNF1α, HNF4α) leading to changes in nutrient flux into metabolic pathways which converge on insulin secretion. Lastly, a large number of genetic syndromes are now linked to hyperinsulinaemic hypoglycaemia. As the molecular and genetic basis of CHI has expanded over the last few years, this review aims to provide an up-to-date knowledge on the genetic causes of CHI.
Keywords: genetics; hyperinsulinism; hypoglycaemia; molecular mechanisms; mutation.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous