

Transmission Trees on a Known Pathogen Phylogeny: Enumeration and Sampling
- PMID: 30873529
- PMCID: PMC6526902
- DOI: 10.1093/molbev/msz058
Transmission Trees on a Known Pathogen Phylogeny: Enumeration and Sampling
Abstract
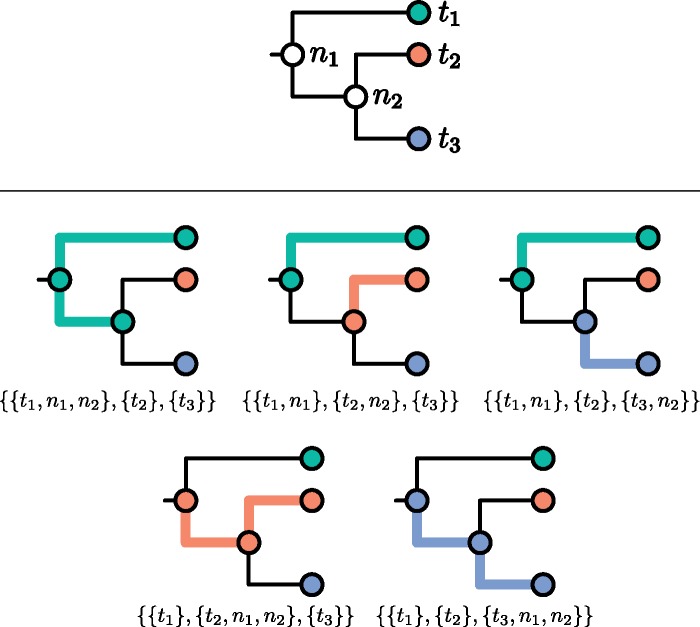
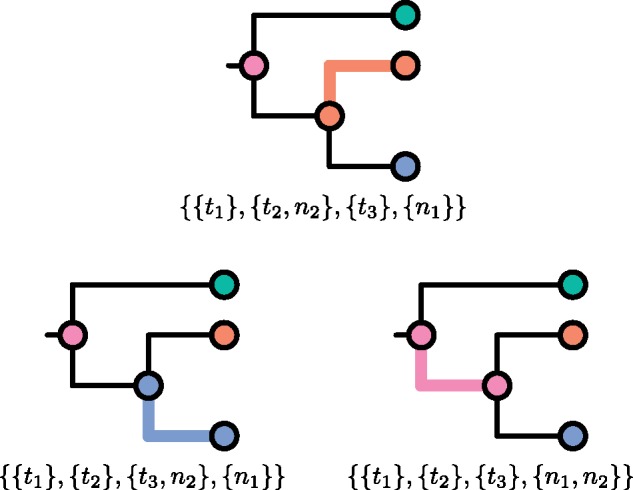
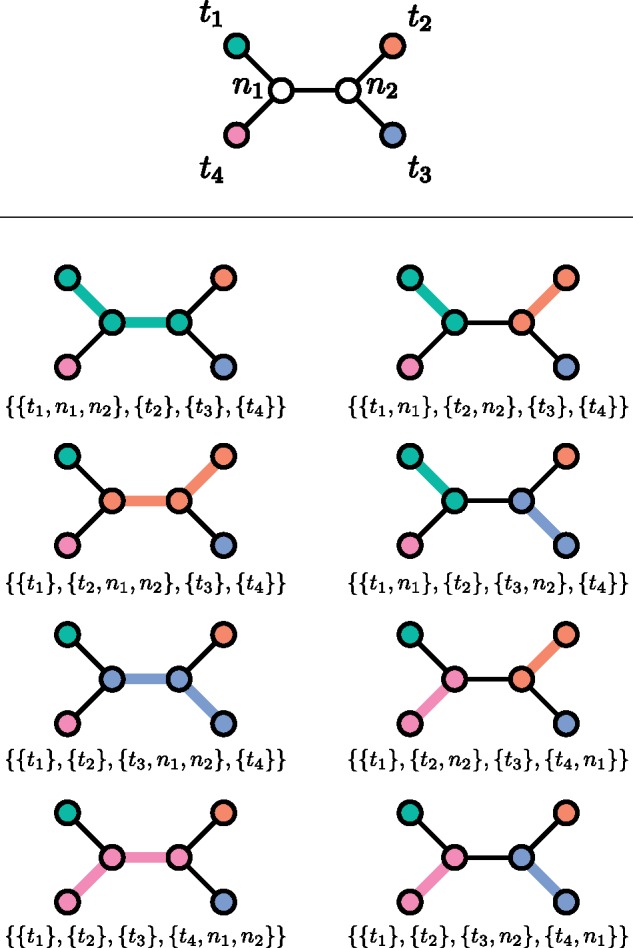
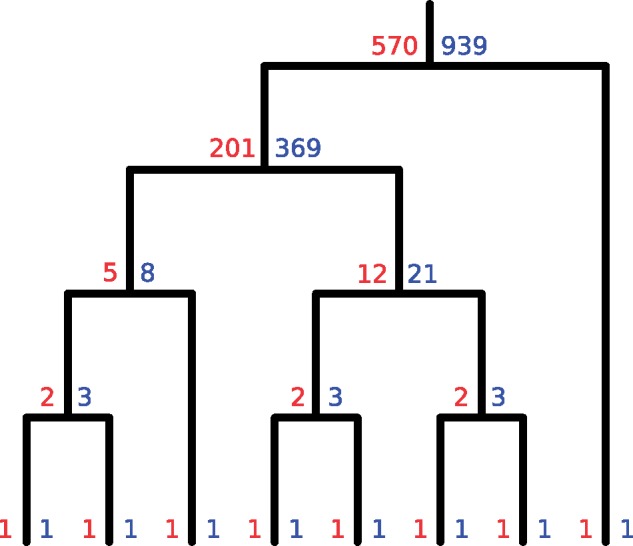
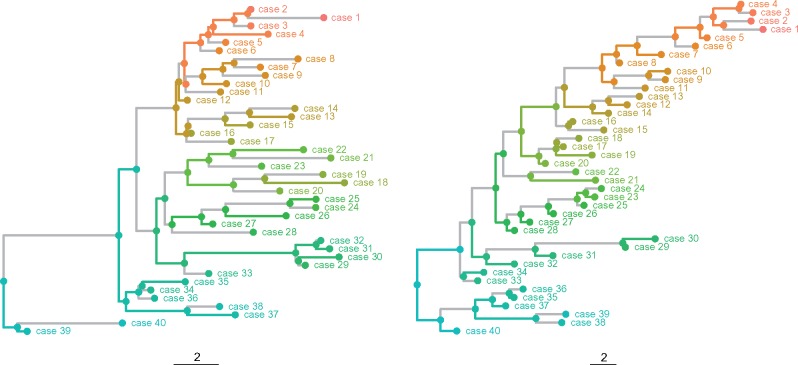
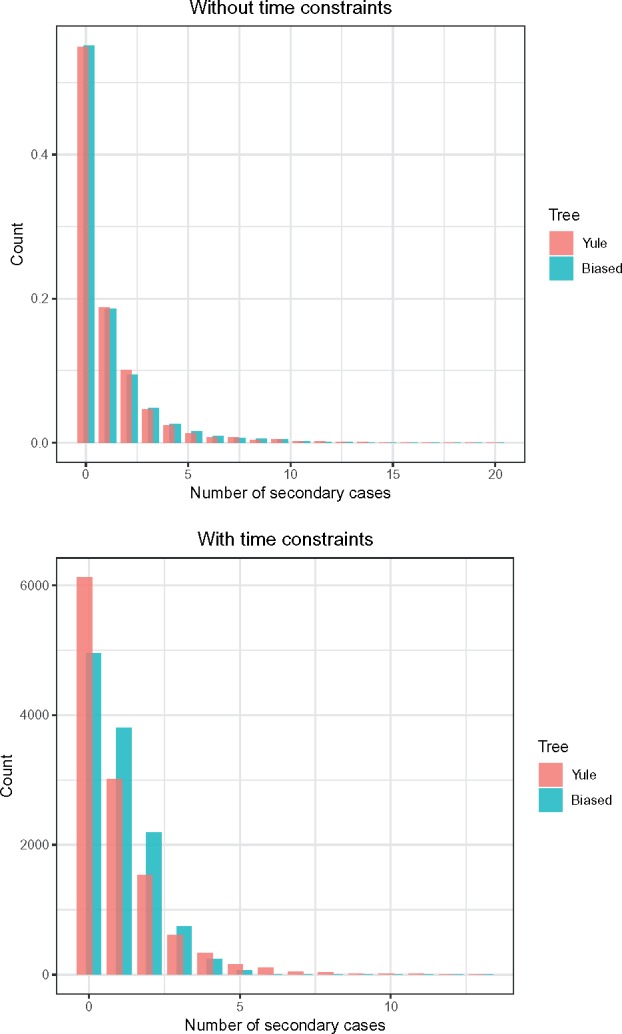
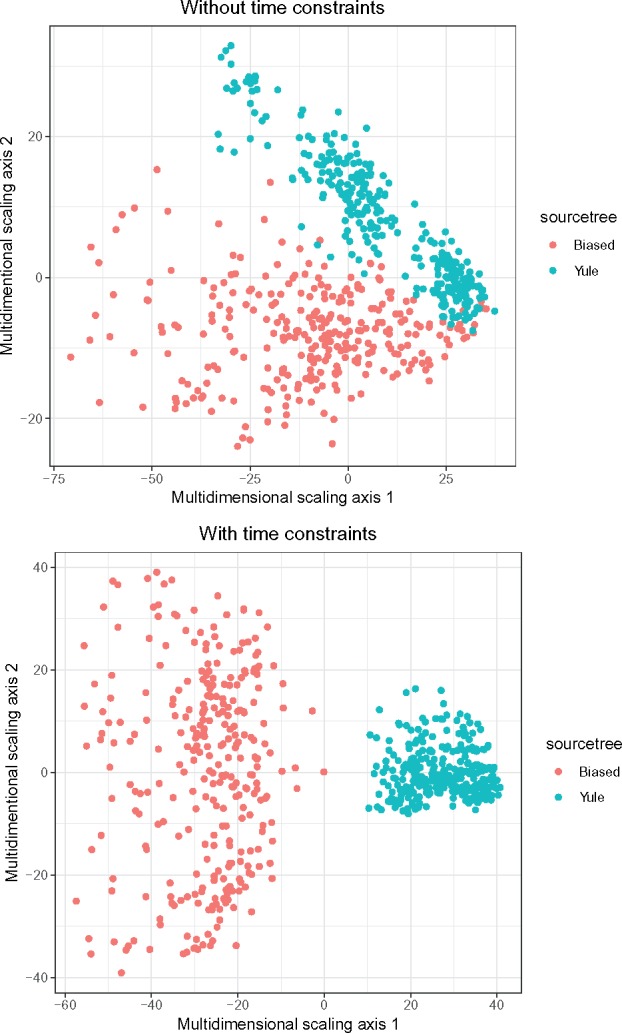
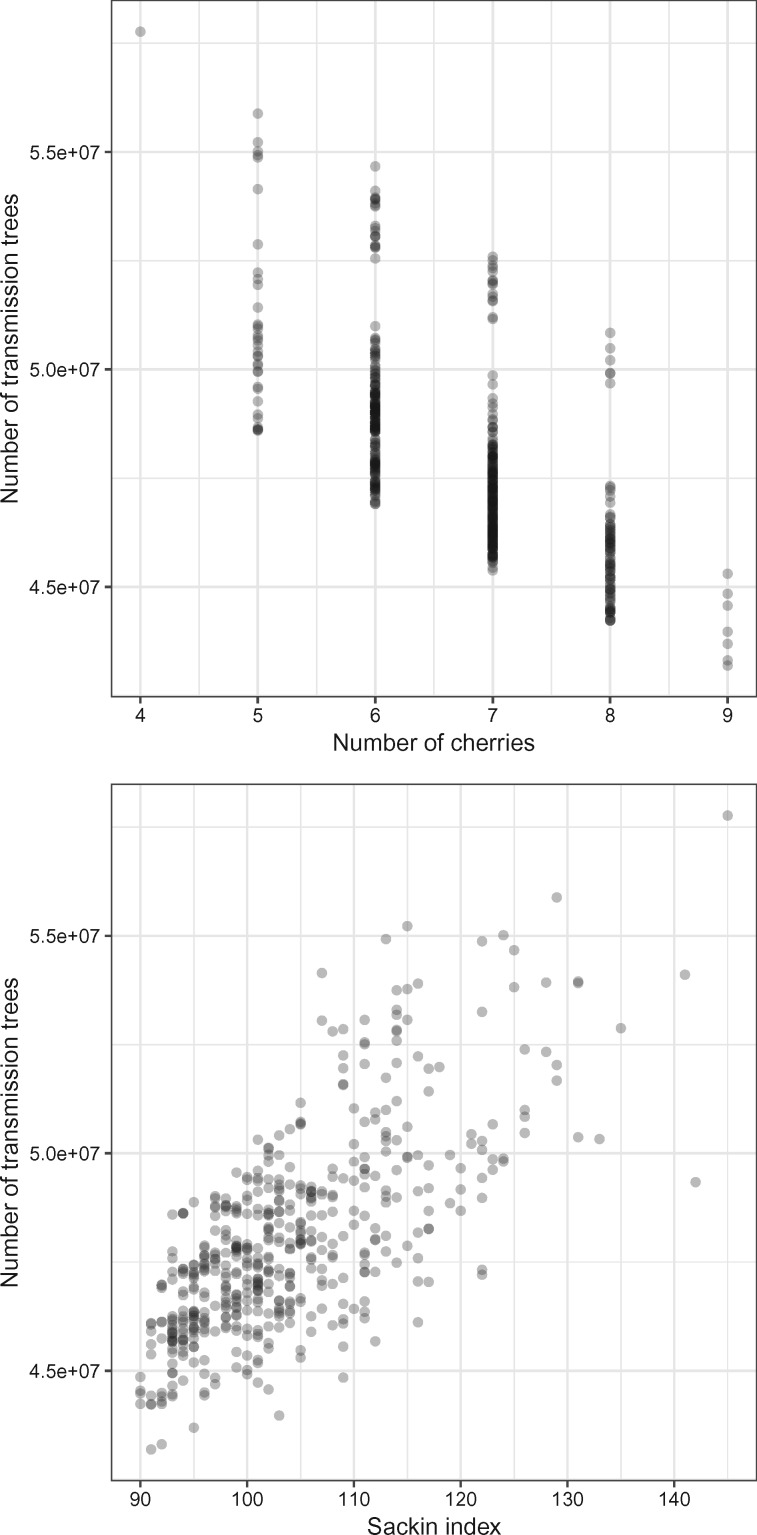
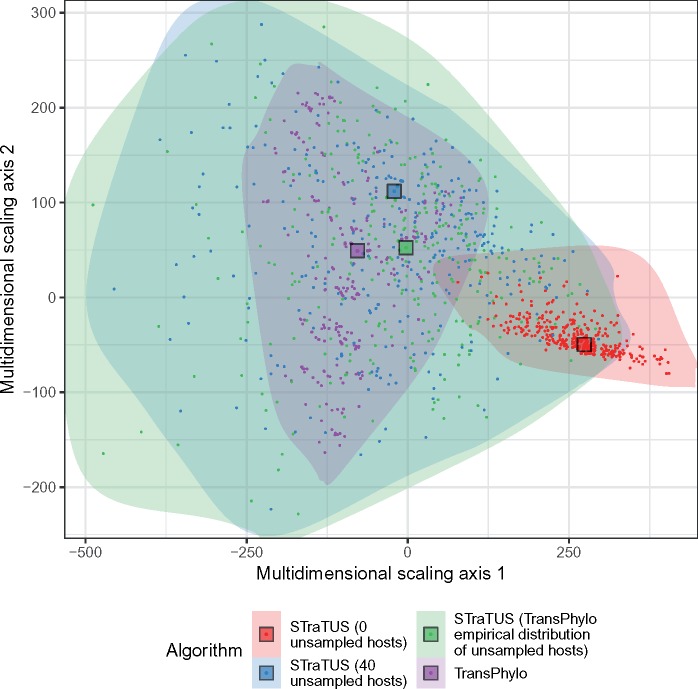
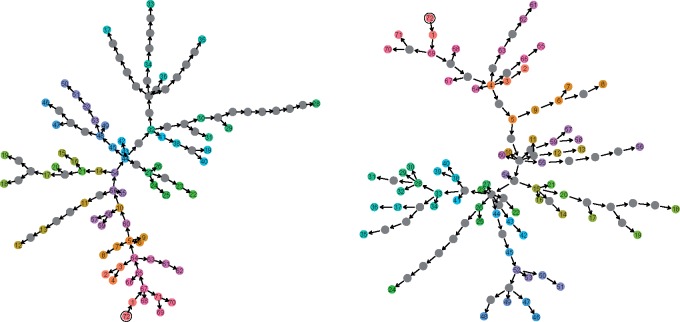
One approach to the reconstruction of infectious disease transmission trees from pathogen genomic data has been to use a phylogenetic tree, reconstructed from pathogen sequences, and annotate its internal nodes to provide a reconstruction of which host each lineage was in at each point in time. If only one pathogen lineage can be transmitted to a new host (i.e., the transmission bottleneck is complete), this corresponds to partitioning the nodes of the phylogeny into connected regions, each of which represents evolution in an individual host. These partitions define the possible transmission trees that are consistent with a given phylogenetic tree. However, the mathematical properties of the transmission trees given a phylogeny remain largely unexplored. Here, we describe a procedure to calculate the number of possible transmission trees for a given phylogeny, and we then show how to uniformly sample from these transmission trees. The procedure is outlined for situations where one sample is available from each host and trees do not have branch lengths, and we also provide extensions for incomplete sampling, multiple sampling, and the application to time trees in a situation where limits on the period during which each host could have been infected and infectious are known. The sampling algorithm is available as an R package (STraTUS).
Keywords: epidemic reconstruction; molecular epidemiology; pathogen genomics; phylogenetics.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Aldrin M, Lyngstad TM, Kristoffersen AB, Storvik B, Borgan Ø, Jansen PA.. 2011. Modelling the spread of infectious salmon anaemia among salmon farms based on seaway distances between farms and genetic relationships between infectious salmon anaemia virus isolates. J R Soc Interface 862: 1346–1356. - PMC - PubMed
-
- Blum MGB, François O.. 2005. On statistical tests of phylogenetic tree imbalance: the sackin and other indices revisited. Math Biosci. 1952: 141–153. - PubMed
-
- Bortolussi N, Durand E, Blum M, François O.. 2006. apTreeshape: statistical analysis of phylogenetic tree shape. Bioinformatics 223: 363–364. - PubMed
-
- Chessel D, Dufour A-B, Thioulouse J.. 2004. The ade4 package – I: one-table methods. R News 41: 5–10.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
