

Germinal center-derived lymphomas: The darkest side of humoral immunity
- PMID: 30874354
- PMCID: PMC6518944
- DOI: 10.1111/imr.12755
Germinal center-derived lymphomas: The darkest side of humoral immunity
Abstract
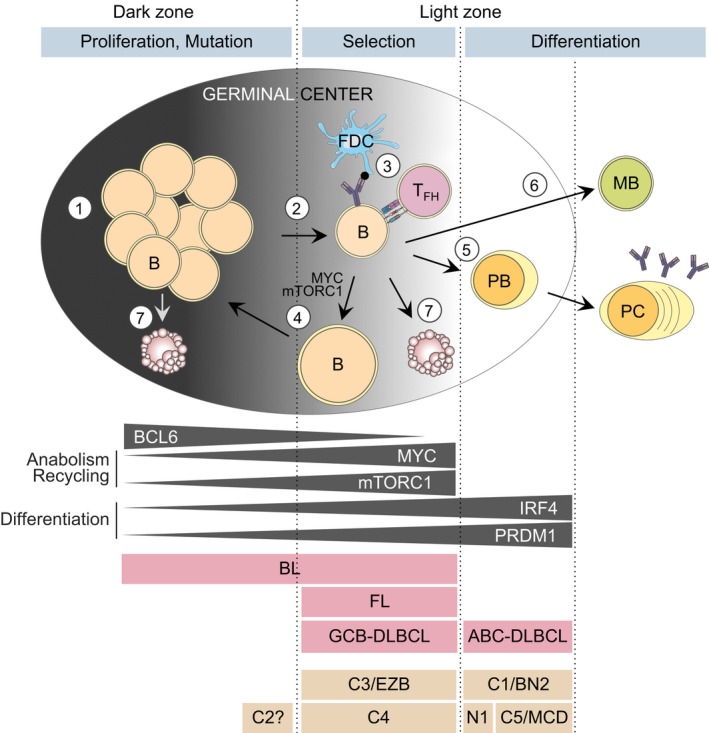
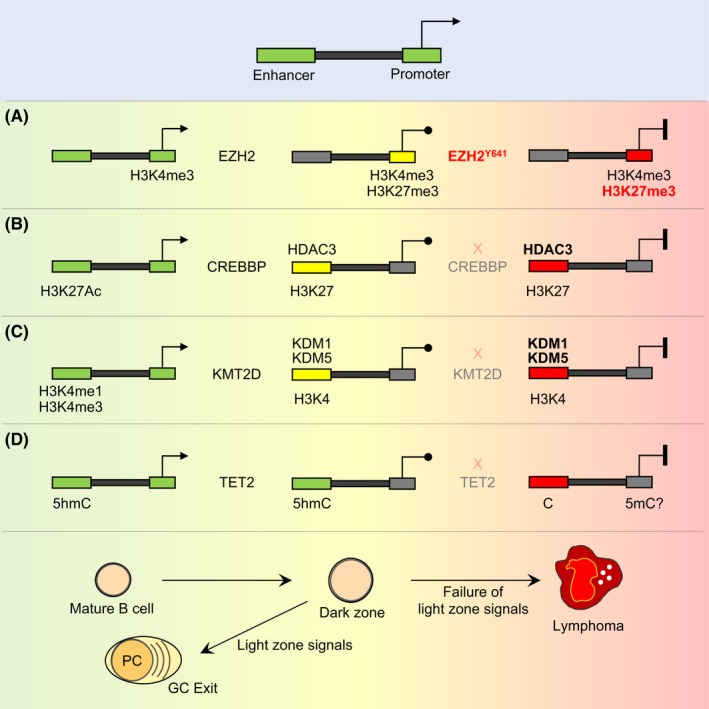
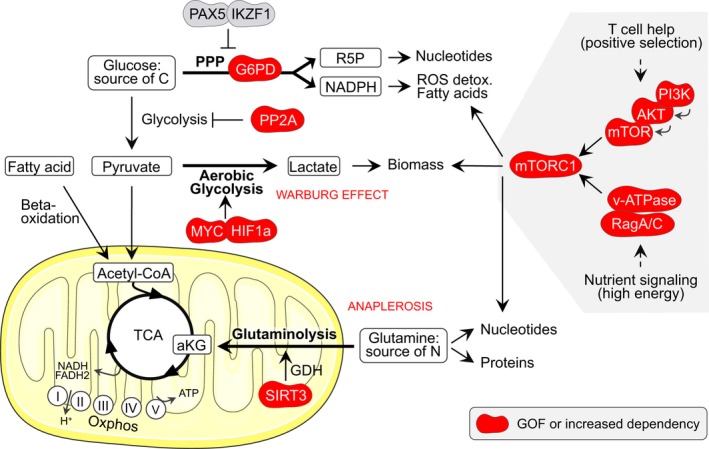
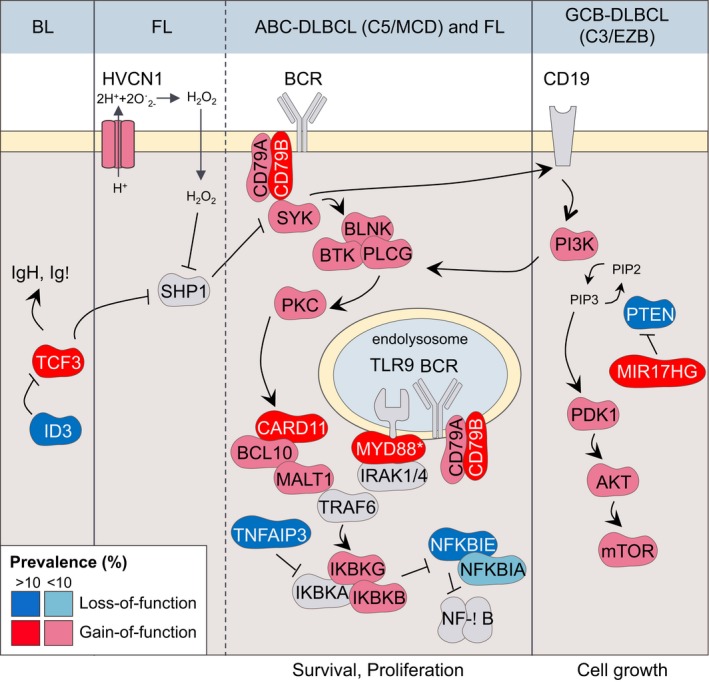
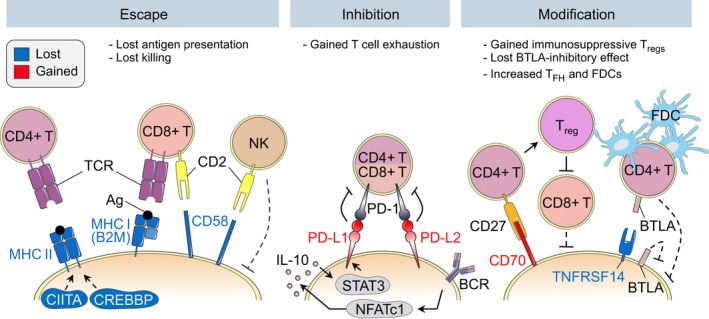
One of the unusual features of germinal center (GC) B cells is that they manifest many hallmarks of cancer cells. Accordingly, most B-cell neoplasms originate from the GC reaction, and characteristically display abundant point mutations, structural genomic lesions, and clonal diversity from the genetic and epigenetic standpoints. The dominant biological theme of GC-derived lymphomas is mutation of genes involved in epigenetic regulation and immune receptor signaling, which come into play at critical transitional stages of the GC reaction. Hence, mechanistic studies of these mutations reveal fundamental insight into the biology of the normal and malignant GC B cell. The BCL6 transcription factor plays a central role in establishing the GC phenotype in B cells, and most lymphomas are dependent on BCL6 to maintain survival, proliferation, and perhaps immune evasion. Many lymphoma mutations have the commonality of enhancing the oncogenic functions of BCL6, or overcoming some of its tumor suppressive effects. Herein, we discuss how unique features of the GC reaction create vulnerabilities that select for particular lymphoma mutations. We examine the interplay between epigenetic programming, metabolism, signaling, and immune regulatory mechanisms in lymphoma, and discuss how these are leading to novel precision therapy strategies to treat lymphoma patients.
Keywords: epigenetic deregulation; germinal center; immune surveillance; lymphomagenesis; precision therapy; signal transduction.
© 2019 The Authors. Immunological Reviews Published by John Wiley & Sons Ltd.
Conflict of interest statement
AM receives research support and has consulted for Janssen Pharmaceutical. He is also a scientific advisor to KDAC pharmaceuticals.
Figures
References
-
- Al‐Tourah AJ, Gill KK, Chhanabhai M, et al. Population‐based analysis of incidence and outcome of transformed non‐Hodgkin's lymphoma. J Clin Oncol. 2008;26(32):5165‐5169. - PubMed
-
- Montoto S, Davies AJ, Matthews J, et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B‐cell lymphoma. J Clin Oncol. 2007;25(17):2426‐2433. - PubMed
-
- Link BK, Maurer MJ, Nowakowski GS, et al. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol. 2013;31(26):3272‐3278. - PMC - PubMed
-
- Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B‐cell lymphomas. N Engl J Med. 1999;341(20):1520‐1529. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
