

Loss of DPP6 in neurodegenerative dementia: a genetic player in the dysfunction of neuronal excitability
- PMID: 30874922
- PMCID: PMC6531610
- DOI: 10.1007/s00401-019-01976-3
Loss of DPP6 in neurodegenerative dementia: a genetic player in the dysfunction of neuronal excitability
Abstract
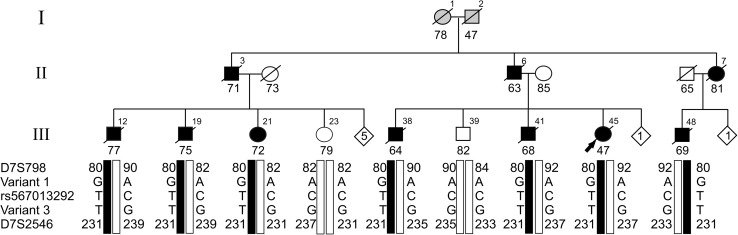
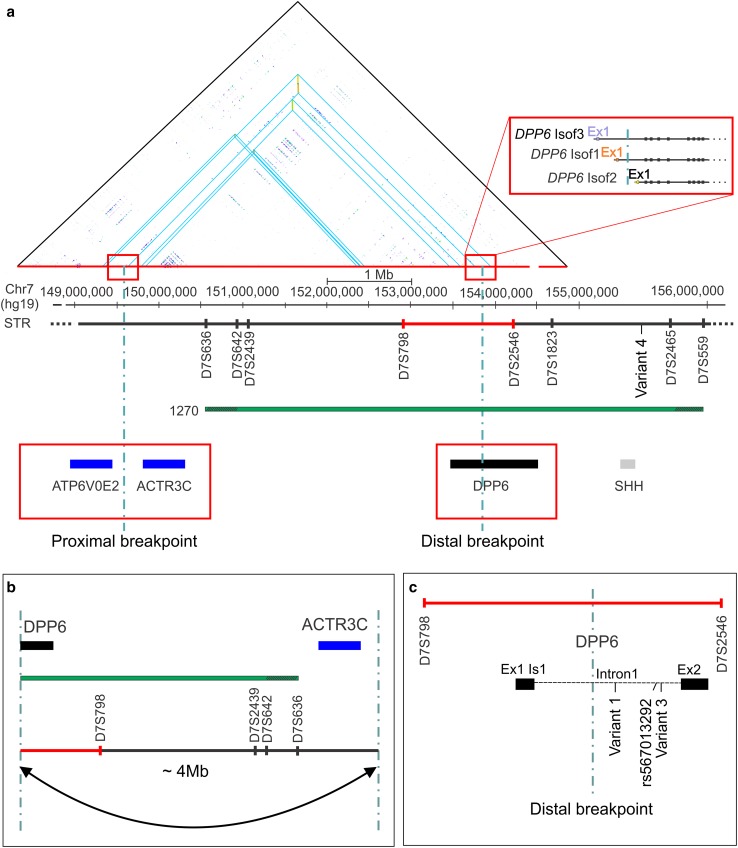
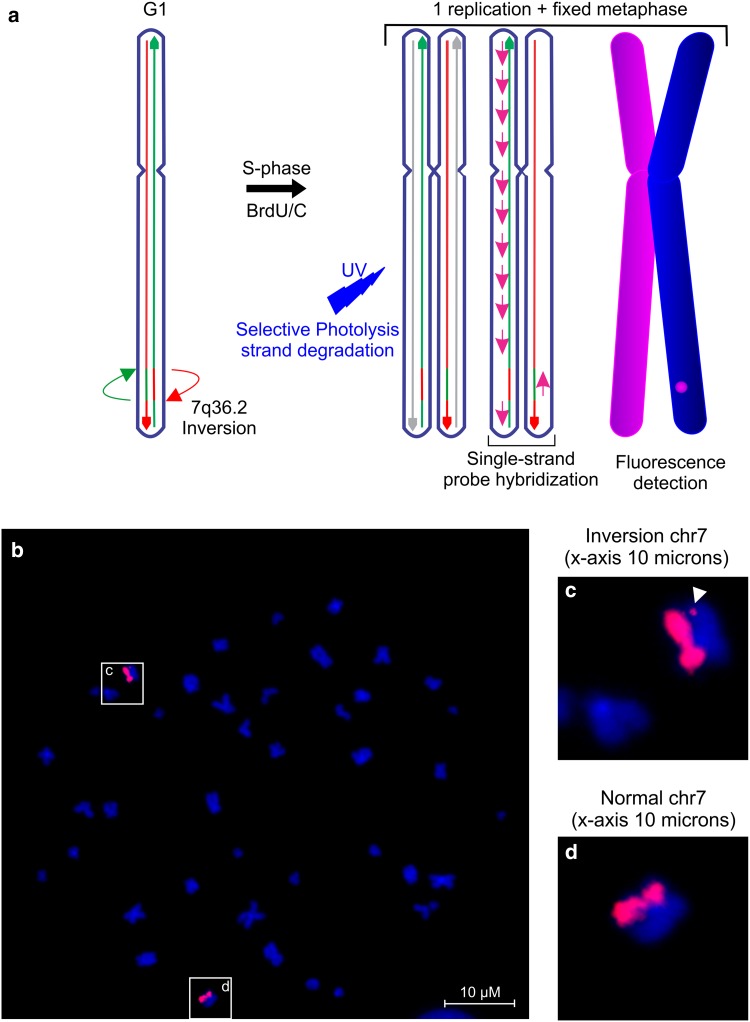
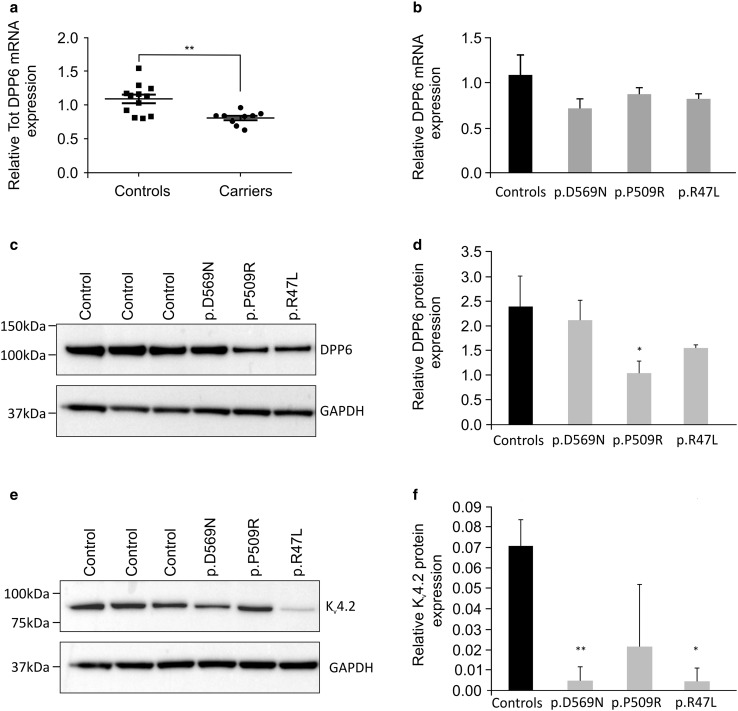
Emerging evidence suggested a converging mechanism in neurodegenerative brain diseases (NBD) involving early neuronal network dysfunctions and alterations in the homeostasis of neuronal firing as culprits of neurodegeneration. In this study, we used paired-end short-read and direct long-read whole genome sequencing to investigate an unresolved autosomal dominant dementia family significantly linked to 7q36. We identified and validated a chromosomal inversion of ca. 4 Mb, segregating on the disease haplotype and disrupting the coding sequence of dipeptidyl-peptidase 6 gene (DPP6). DPP6 resequencing identified significantly more rare variants-nonsense, frameshift, and missense-in early-onset Alzheimer's disease (EOAD, p value = 0.03, OR = 2.21 95% CI 1.05-4.82) and frontotemporal dementia (FTD, p = 0.006, OR = 2.59, 95% CI 1.28-5.49) patient cohorts. DPP6 is a type II transmembrane protein with a highly structured extracellular domain and is mainly expressed in brain, where it binds to the potassium channel Kv4.2 enhancing its expression, regulating its gating properties and controlling the dendritic excitability of hippocampal neurons. Using in vitro modeling, we showed that the missense variants found in patients destabilize DPP6 and reduce its membrane expression (p < 0.001 and p < 0.0001) leading to a loss of protein. Reduced DPP6 and/or Kv4.2 expression was also detected in brain tissue of missense variant carriers. Loss of DPP6 is known to cause neuronal hyperexcitability and behavioral alterations in Dpp6-KO mice. Taken together, the results of our genomic, genetic, expression and modeling analyses, provided direct evidence supporting the involvement of DPP6 loss in dementia. We propose that loss of function variants have a higher penetrance and disease impact, whereas the missense variants have a variable risk contribution to disease that can vary from high to low penetrance. Our findings of DPP6, as novel gene in dementia, strengthen the involvement of neuronal hyperexcitability and alteration in the homeostasis of neuronal firing as a disease mechanism to further investigate.
Keywords: DPP6; Dementia; Hyperexcitability; Neurodegenerative brain diseases; Oxford nanopore technologies (ONT) PromethION; Whole genome sequencing.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
-
- Aronesty E. Comparison of sequencing utility programs. Open Bioinform J. 2013;7:1–8. doi: 10.2174/1875036201307010001. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
