

Hyperhomocysteinemia leads to exacerbation of ischemic brain damage: Role of GluN2A NMDA receptors
- PMID: 30885791
- PMCID: PMC6588434
- DOI: 10.1016/j.nbd.2019.03.012
Hyperhomocysteinemia leads to exacerbation of ischemic brain damage: Role of GluN2A NMDA receptors
Abstract
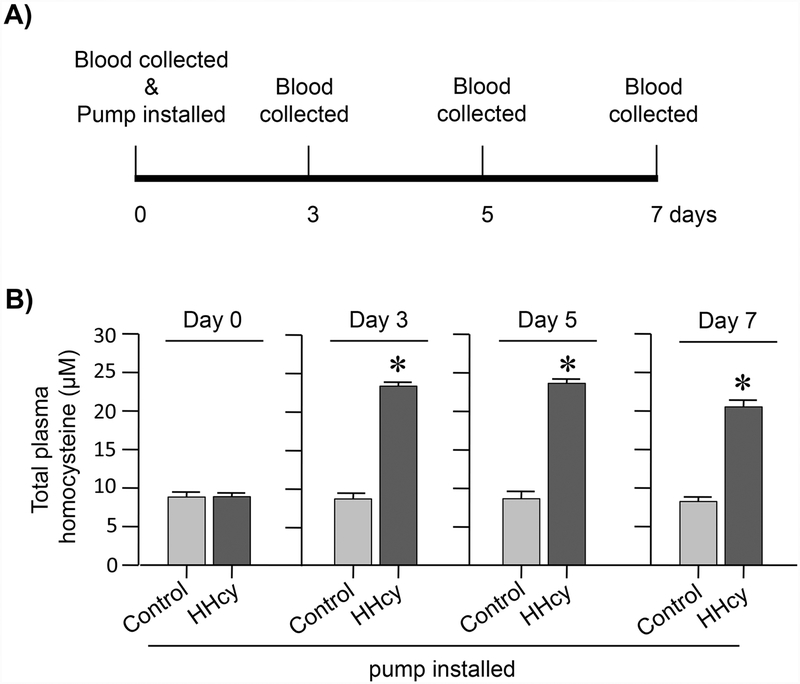
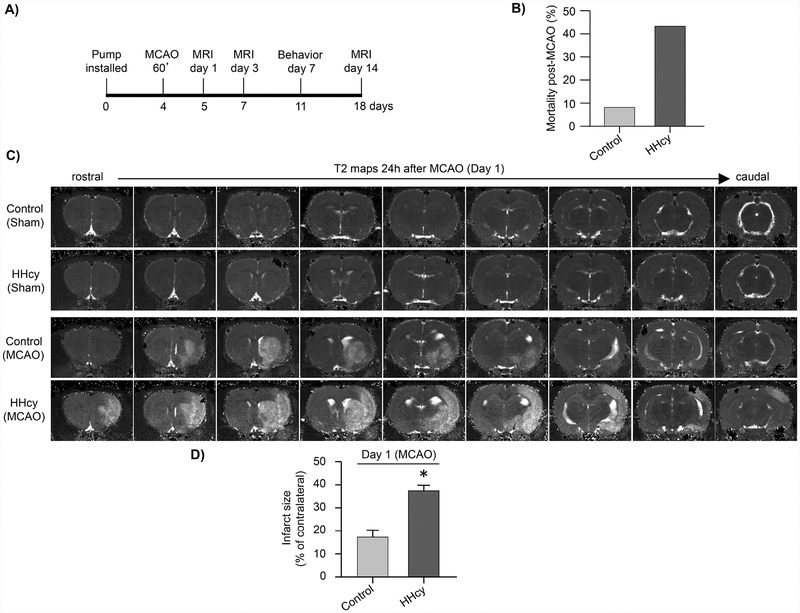
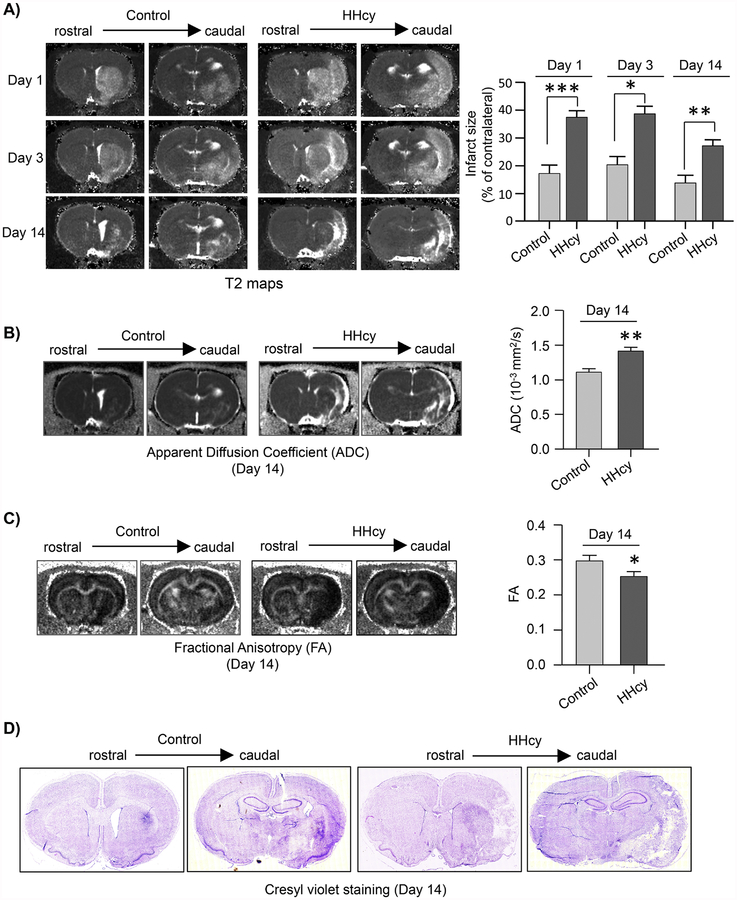
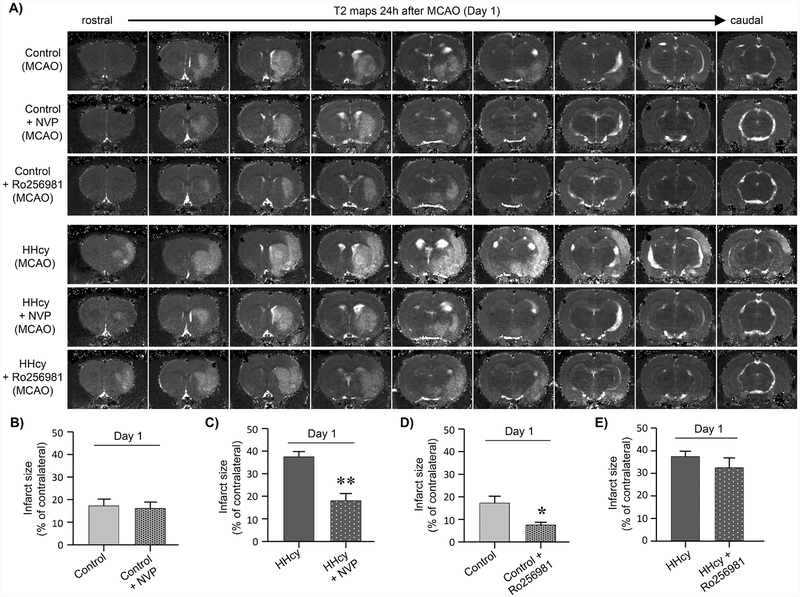
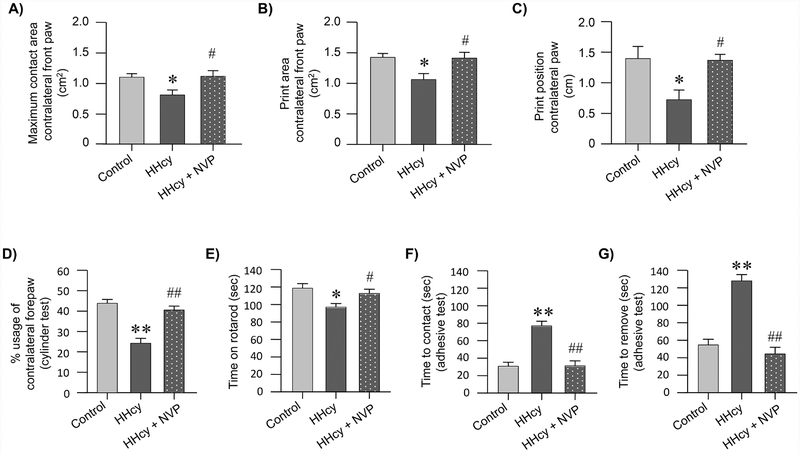
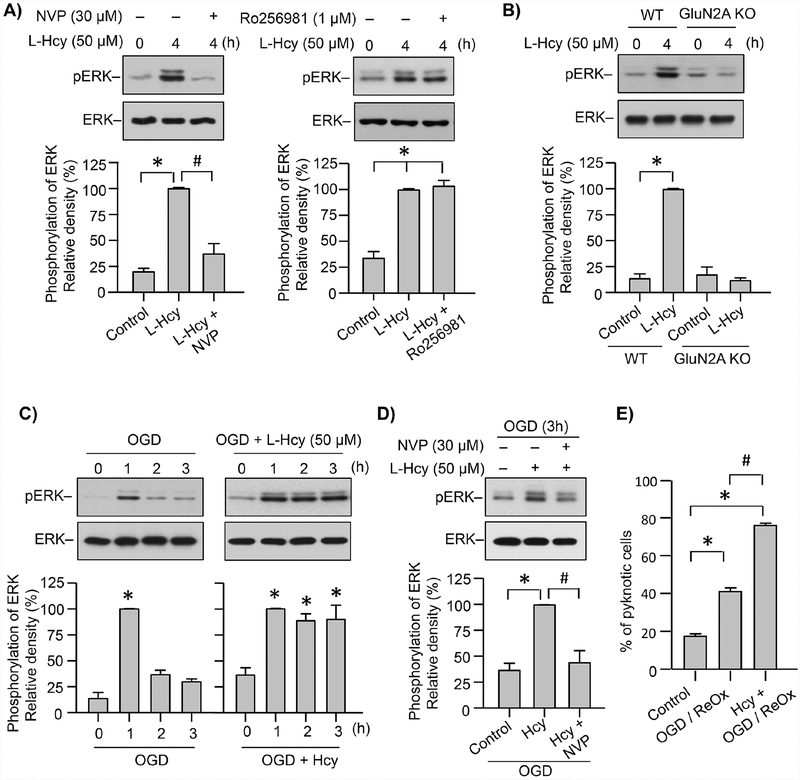
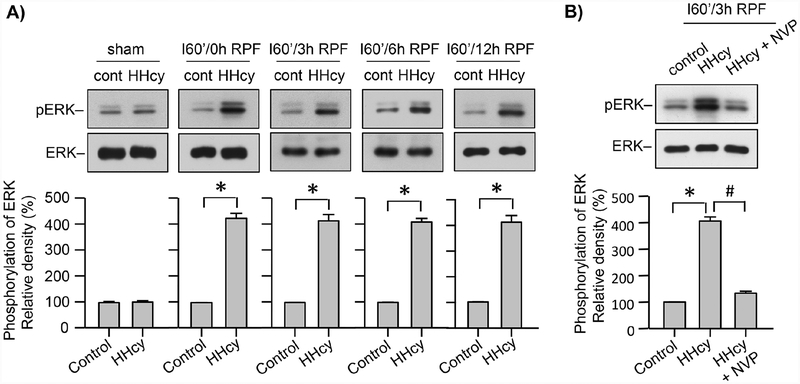
Hyperhomocysteinemia has been implicated in several neurodegenerative disorders including ischemic stroke. However, the pathological consequences of ischemic insult in individuals predisposed to hyperhomocysteinemia and the associated etiology are unknown. In this study, we evaluated the outcome of transient ischemic stroke in a rodent model of hyperhomocysteinemia, developed by subcutaneous implantation of osmotic pumps containing L-homocysteine into male Wistar rats. Our findings show a 42.3% mortality rate in hyperhomocysteinemic rats as compared to 7.7% in control rats. Magnetic resonance imaging of the brain in the surviving rats shows that mild hyperhomocysteinemia leads to exacerbation of ischemic injury within 24 h, which remains elevated over time. Behavioral studies further demonstrate significant deficit in sensorimotor functions in hyperhomocysteinemic rats compared to control rats. Using pharmacological inhibitors targeting the NMDAR subtypes, the study further demonstrates that inhibition of GluN2A-containing NMDARs significantly reduces ischemic brain damage in hyperhomocysteinemic rats but not in control rats, indicating that hyperhomocysteinemia-mediated exacerbation of ischemic brain injury involves GluN2A-NMDAR signaling. Complementary studies in GluN2A-knockout mice show that in the absence of GluN2A-NMDARs, hyperhomocysteinemia-associated exacerbation of ischemic brain injury is blocked, confirming that GluN2A-NMDAR activation is a critical determinant of the severity of ischemic damage under hyperhomocysteinemic conditions. Furthermore, at the molecular level we observe GluN2A-NMDAR dependent sustained increase in ERK MAPK phosphorylation under hyperhomocysteinemic condition that has been shown to be involved in homocysteine-induced neurotoxicity. Taken together, the findings show that hyperhomocysteinemia triggers a unique signaling pathway that in conjunction with ischemia-induced pathways enhance the pathology of stroke under hyperhomocysteinemic conditions.
Keywords: Behavioral studies; ERK MAPK; GluN2A-NMDA receptor knockout mice; GluN2A-NMDA receptors (also known as NR2A-NMDA receptors); GluN2B-NMDA receptors (also known as NR2B-NMDA receptors); Hyperhomocysteinemia; Ischemic brain injury; Magnetic resonance imaging; Middle cerebral artery occlusion.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
CONFLICT OF INTEREST:
The authors declare no competing financial interests.
Figures


Similar articles
-
Hyperhomocysteinemia is an emerging comorbidity in ischemic stroke.Exp Neurol. 2021 Feb;336:113541. doi: 10.1016/j.expneurol.2020.113541. Epub 2020 Dec 3. Exp Neurol. 2021. PMID: 33278453 Free PMC article. Review.
-
GluN2A-NMDA receptor-mediated sustained Ca2+ influx leads to homocysteine-induced neuronal cell death.J Biol Chem. 2019 Jul 19;294(29):11154-11165. doi: 10.1074/jbc.RA119.008820. Epub 2019 Jun 5. J Biol Chem. 2019. PMID: 31167782 Free PMC article.
-
Role of GluN2A NMDA receptor in homocysteine-induced prostaglandin E2 release from neurons.J Neurochem. 2019 Jul;150(1):44-55. doi: 10.1111/jnc.14775. Epub 2019 Jun 20. J Neurochem. 2019. PMID: 31125437 Free PMC article.
-
Homocysteine-induced sustained GluN2A NMDA receptor stimulation leads to mitochondrial ROS generation and neurotoxicity.J Biol Chem. 2024 May;300(5):107253. doi: 10.1016/j.jbc.2024.107253. Epub 2024 Apr 1. J Biol Chem. 2024. PMID: 38569938 Free PMC article.
-
The Role of GluN2A in Cerebral Ischemia: Promoting Neuron Death and Survival in the Early Stage and Thereafter.Mol Neurobiol. 2018 Feb;55(2):1208-1216. doi: 10.1007/s12035-017-0395-8. Epub 2017 Jan 19. Mol Neurobiol. 2018. PMID: 28102473 Review.
Cited by
-
Hyperhomocysteinemia is an emerging comorbidity in ischemic stroke.Exp Neurol. 2021 Feb;336:113541. doi: 10.1016/j.expneurol.2020.113541. Epub 2020 Dec 3. Exp Neurol. 2021. PMID: 33278453 Free PMC article. Review.
-
Homocysteine enhances the excitability of cultured hippocampal neurons without altering the gene expression of voltage-gated ion channels.Mol Brain. 2025 Apr 10;18(1):31. doi: 10.1186/s13041-025-01205-x. Mol Brain. 2025. PMID: 40211242 Free PMC article.
-
N-Methyl-D-aspartate receptor activation, novel mechanism of homocysteine-induced blood-retinal barrier dysfunction.J Mol Med (Berl). 2021 Jan;99(1):119-130. doi: 10.1007/s00109-020-02000-y. Epub 2020 Nov 6. J Mol Med (Berl). 2021. PMID: 33159240 Free PMC article.
-
Influence of the brain‑gut axis on neuroinflammation in cerebral ischemia‑reperfusion injury (Review).Int J Mol Med. 2024 Mar;53(3):30. doi: 10.3892/ijmm.2024.5354. Epub 2024 Feb 1. Int J Mol Med. 2024. PMID: 38299236 Free PMC article. Review.
-
The Role of Methyl Donors of the Methionine Cycle in Gastrointestinal Infection and Inflammation.Healthcare (Basel). 2021 Dec 29;10(1):61. doi: 10.3390/healthcare10010061. Healthcare (Basel). 2021. PMID: 35052225 Free PMC article. Review.
References
-
- Auberson YP, et al., 2002. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 12, 1099–102. - PubMed
-
- Austin RC, et al., 2004. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 11 Suppl 1, S56–64. - PubMed
-
- Barria A, Malinow R, 2002. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 35, 345–53. - PubMed
-
- Bouet V, et al., 2009. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 4, 1560–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
