

Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex
- PMID: 30885957
- PMCID: PMC6478521
- DOI: 10.4049/jimmunol.1801627
Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex
Abstract
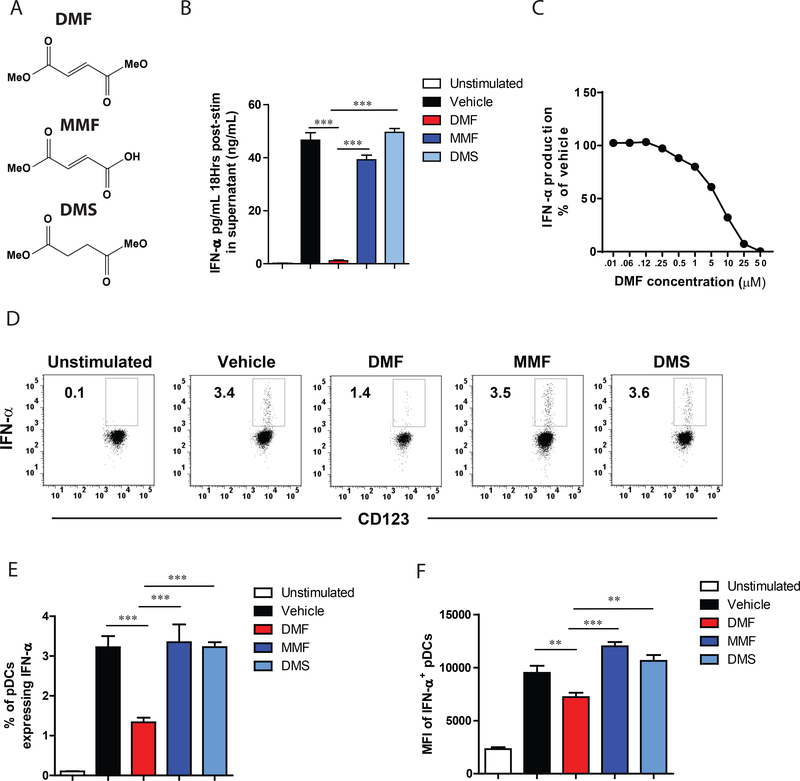
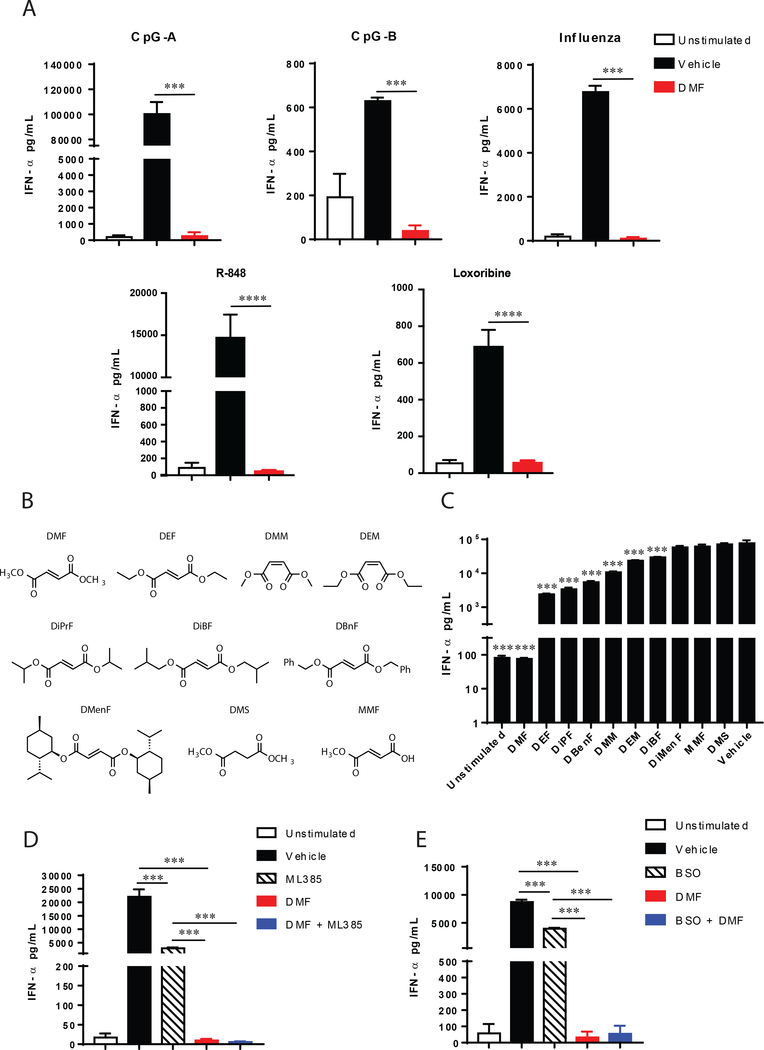
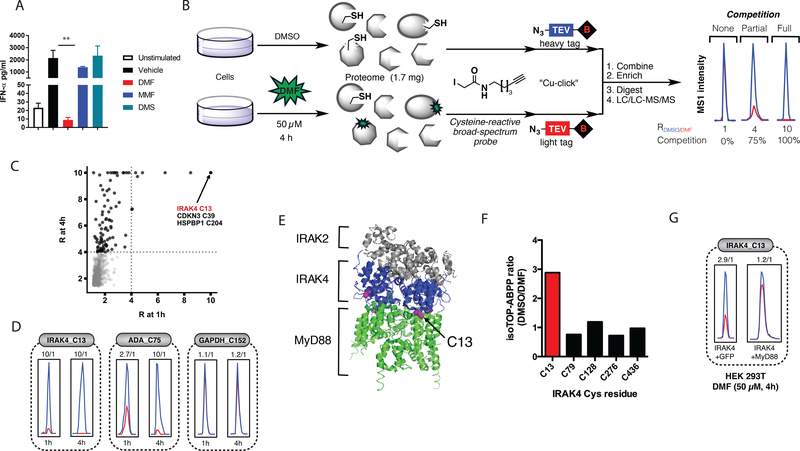
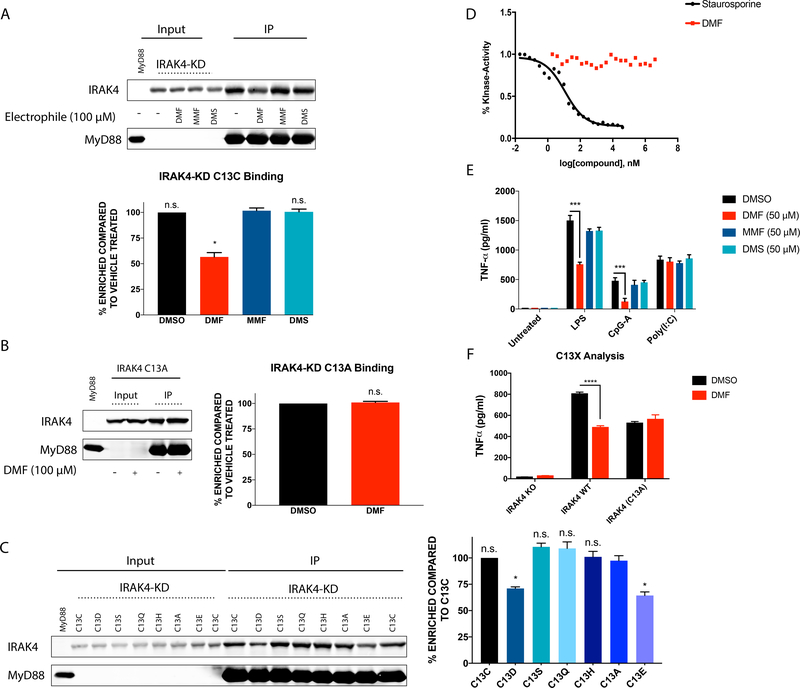
Dimethyl fumarate (DMF) is a prescribed treatment for multiple sclerosis and has also been used to treat psoriasis. The electrophilicity of DMF suggests that its immunosuppressive activity is related to the covalent modification of cysteine residues in the human proteome. Nonetheless, our understanding of the proteins modified by DMF in human immune cells and the functional consequences of these reactions remains incomplete. In this study, we report that DMF inhibits human plasmacytoid dendritic cell function through a mechanism of action that is independent of the major electrophile sensor NRF2. Using chemical proteomics, we instead identify cysteine 13 of the innate immune kinase IRAK4 as a principal cellular target of DMF. We show that DMF blocks IRAK4-MyD88 interactions and IRAK4-mediated cytokine production in a cysteine 13-dependent manner. Our studies thus identify a proteomic hotspot for DMF action that constitutes a druggable protein-protein interface crucial for initiating innate immune responses.
Copyright © 2019 by The American Association of Immunologists, Inc.
Figures
Similar articles
-
A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity.J Exp Med. 2007 May 14;204(5):1025-36. doi: 10.1084/jem.20061825. Epub 2007 Apr 30. J Exp Med. 2007. PMID: 17470642 Free PMC article.
-
Dimethyl fumarate: Regulatory effects on the immune system in the treatment of multiple sclerosis.J Cell Physiol. 2019 Jul;234(7):9943-9955. doi: 10.1002/jcp.27930. Epub 2018 Dec 7. J Cell Physiol. 2019. PMID: 30536402 Review.
-
Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells.Sci Signal. 2016 Sep 13;9(445):rs10. doi: 10.1126/scisignal.aaf7694. Sci Signal. 2016. PMID: 27625306 Free PMC article.
-
Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro.PLoS One. 2015 Mar 20;10(3):e0120254. doi: 10.1371/journal.pone.0120254. eCollection 2015. PLoS One. 2015. PMID: 25793262 Free PMC article.
-
Dimethyl fumarate downregulates the immune response through the HCA2/GPR109A pathway: Implications for the treatment of multiple sclerosis.Mult Scler Relat Disord. 2018 Jul;23:46-50. doi: 10.1016/j.msard.2018.04.016. Epub 2018 Apr 25. Mult Scler Relat Disord. 2018. PMID: 29763776 Review.
Cited by
-
Dichloro Butenediamides as Irreversible Site-Selective Protein Conjugation Reagent.Angew Chem Int Ed Engl. 2021 Oct 25;60(44):23750-23755. doi: 10.1002/anie.202108791. Epub 2021 Sep 29. Angew Chem Int Ed Engl. 2021. PMID: 34472678 Free PMC article.
-
AzidoTMT Enables Direct Enrichment and Highly Multiplexed Quantitation of Proteome-Wide Functional Residues.J Proteome Res. 2023 Jul 7;22(7):2218-2231. doi: 10.1021/acs.jproteome.2c00703. Epub 2023 Jun 7. J Proteome Res. 2023. PMID: 37285454 Free PMC article.
-
Wdr1 and cofilin are necessary mediators of immune-cell-specific apoptosis triggered by Tecfidera.Nat Commun. 2021 Sep 30;12(1):5736. doi: 10.1038/s41467-021-25466-x. Nat Commun. 2021. PMID: 34593792 Free PMC article.
-
Innate immune signaling and immunothrombosis: New insights and therapeutic opportunities.Eur J Immunol. 2022 Jul;52(7):1024-1034. doi: 10.1002/eji.202149410. Epub 2022 May 24. Eur J Immunol. 2022. PMID: 35569038 Free PMC article. Review.
-
Dimethyl fumarate ameliorates hepatic inflammation in alcohol related liver disease.Liver Int. 2020 Jul;40(7):1610-1619. doi: 10.1111/liv.14483. Epub 2020 May 6. Liver Int. 2020. PMID: 32306456 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
