

Gene activation precedes DNA demethylation in response to infection in human dendritic cells
- PMID: 30886108
- PMCID: PMC6452747
- DOI: 10.1073/pnas.1814700116
Gene activation precedes DNA demethylation in response to infection in human dendritic cells
Abstract
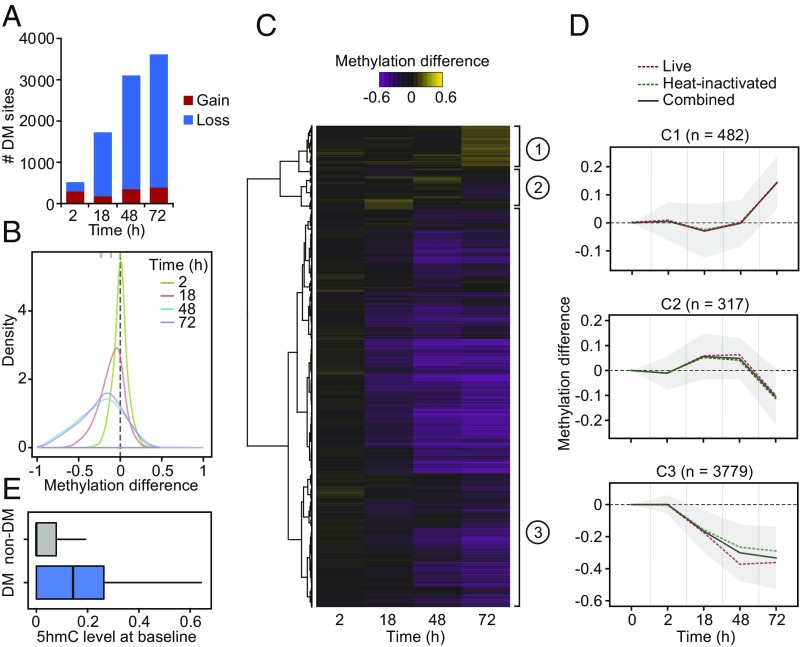
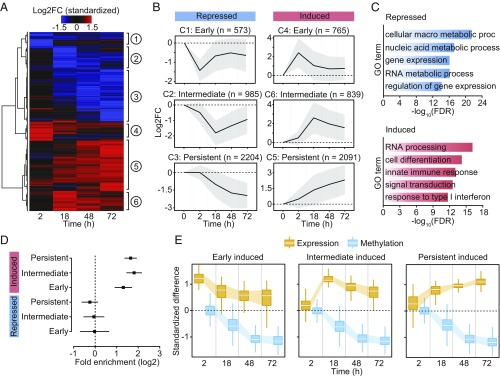
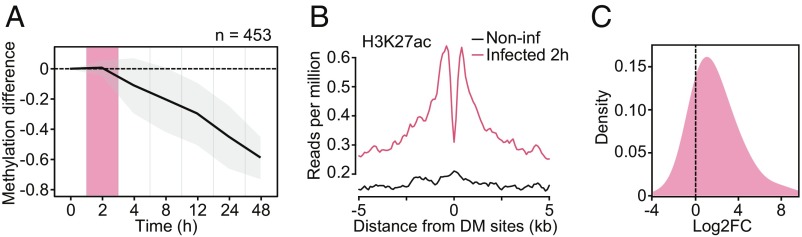
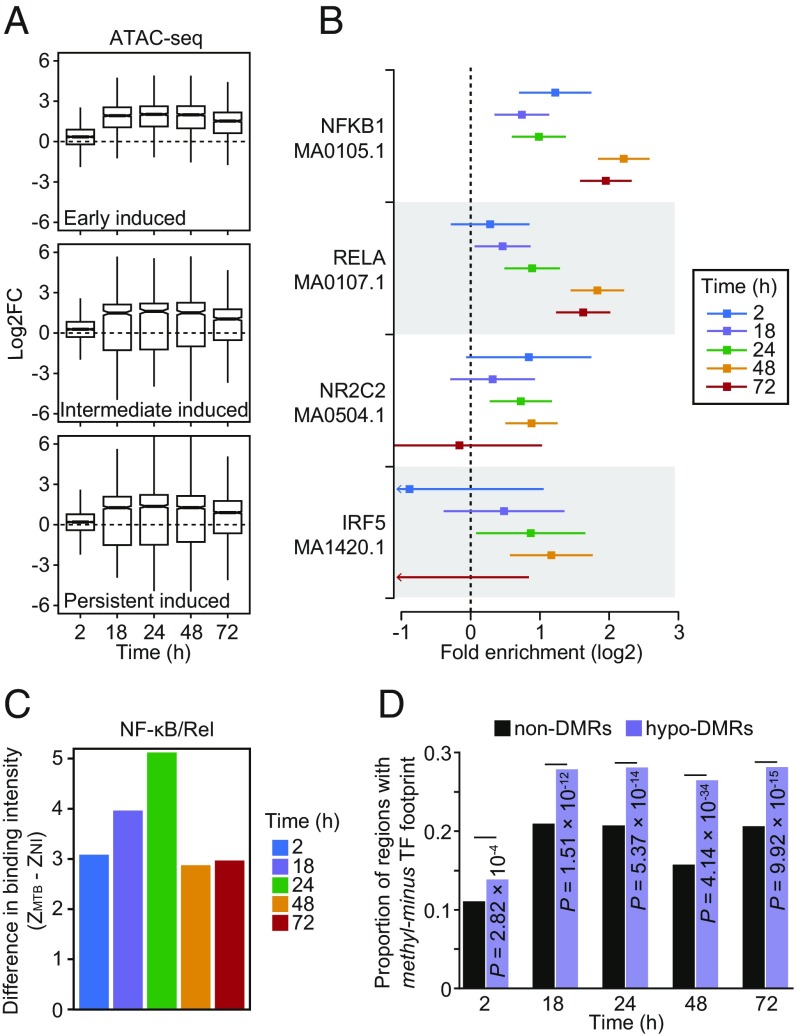
DNA methylation is considered to be a relatively stable epigenetic mark. However, a growing body of evidence indicates that DNA methylation levels can change rapidly; for example, in innate immune cells facing an infectious agent. Nevertheless, the causal relationship between changes in DNA methylation and gene expression during infection remains to be elucidated. Here, we generated time-course data on DNA methylation, gene expression, and chromatin accessibility patterns during infection of human dendritic cells with Mycobacterium tuberculosis We found that the immune response to infection is accompanied by active demethylation of thousands of CpG sites overlapping distal enhancer elements. However, virtually all changes in gene expression in response to infection occur before detectable changes in DNA methylation, indicating that the observed losses in methylation are a downstream consequence of transcriptional activation. Footprinting analysis revealed that immune-related transcription factors (TFs), such as NF-κB/Rel, are recruited to enhancer elements before the observed losses in methylation, suggesting that DNA demethylation is mediated by TF binding to cis-acting elements. Collectively, our results show that DNA demethylation plays a limited role to the establishment of the core regulatory program engaged upon infection.
Keywords: DNA methylation; dendritic cells; epigenetic; immune responses; tuberculosis.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
