

doi: 10.1038/s41588-019-0366-2.
Epub 2019 Mar 18.
Linked-read analysis identifies mutations in single-cell DNA-sequencing data
Affiliations
- PMID: 30886424
- PMCID: PMC6900933
- DOI: 10.1038/s41588-019-0366-2
Item in Clipboard
Linked-read analysis identifies mutations in single-cell DNA-sequencing data
Nat Genet.
2019 Apr.
Abstract
Whole-genome sequencing of DNA from single cells has the potential to reshape our understanding of mutational heterogeneity in normal and diseased tissues. However, a major difficulty is distinguishing amplification artifacts from biologically derived somatic mutations. Here, we describe linked-read analysis (LiRA), a method that accurately identifies somatic single-nucleotide variants (sSNVs) by using read-level phasing with nearby germline heterozygous polymorphisms, thereby enabling the characterization of mutational signatures and estimation of somatic mutation rates in single cells.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures
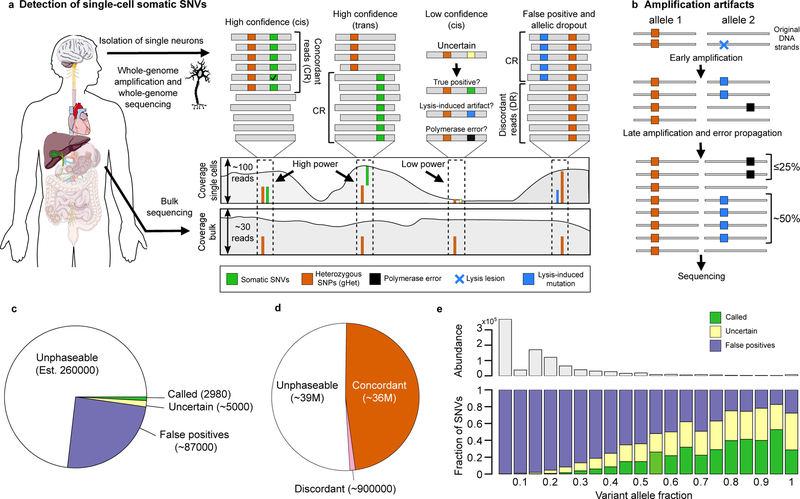
a, Methodology for identifying false positive (FP) somatic SNVs (sSNVs). LiRA analyzes reads and mate-pair reads that cover the positions of an sSNV and a gHet (spanning reads). ‘Concordant’ reads (CR) support the gHet allele (alt/ref in cis/trans) and the sSNV alt call. ‘Discordant reads’ (DR) support the gHet allele but the reference base at the sSNV position. b, Model for how the linked read pattern specific to FPs arises from DNA lesions or polymerase errors. A lesion may be present on and copied from one strand of input DNA (blue), or ϕ29 polymerase may mispair a base (black). Both errors are exponentially amplified. As polymerase errors are introduced after the first round of amplification at the earliest, they are expected to appear in ≤25% of gHet-linked reads, whereas lesion-derived artifacts are expected to appear in ~50%. c, Classification of candidate sSNVs in LiRA. Most sSNV candidates (est. ~260,000, 73%) are too far away from a gHet to be covered by the same read or mate-pair. Over the powered fraction (27%, ~95,000), most (92%, ~87,000) are filtered as false positives due to the presence of at least one discordant read covering the sSNV position and each linked gHet. In the remaining subset, most (63%, ~5,000) do not meet LiRA’s quality thresholds, and 2,980 (37%, 0.8% overall) are reported as LiRA sSNV calls. d, Phasing of gHets. Just under half of gHets are close enough to other gHets to be linked, and only 2% are filtered (erroneously) as false positives. e, Call status of candidate sSNVs in LiRA by variant allele fraction (VAF). Most sSNV candidates are low VAF; LiRA filters almost all low VAF sSNV candidates. As VAF increases, sSNV candidates are more frequently called, but a substantial proportion of high VAF candidates are still false positives.
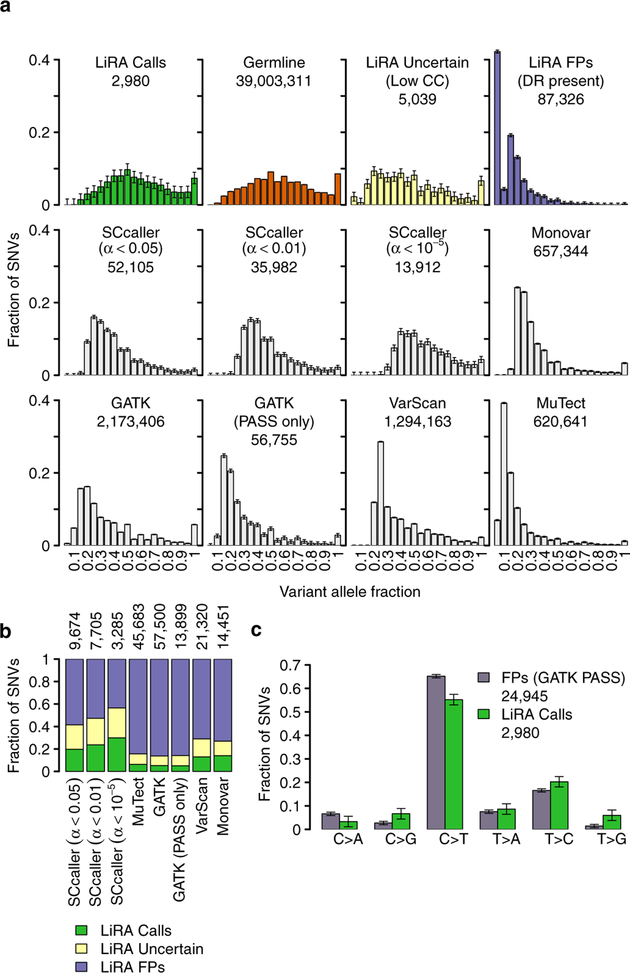
a, Comparison of the variant allele fraction (VAF) of LiRA high-confidence calls, uncertain sSNVs, and FPs to germline mutations and other calling methods. LiRA calls have a VAF distribution indistinguishable from that of heterozygous germline polymorphisms, while LiRA uncertain mutations and FPs are moderately and severely skewed towards low VAF values, respectively. Other single-cell variant calling methods also produce VAF distributions skewed towards low VAF values. Accepting only PASS mutations after VQSR in GATK does not change this. In SCcaller, α is the probability that a candidate sSNV is an amplification artifact, and a set of calls is obtained by accepting only those with α less than a user-set threshold. Lowering α mitigates but does not remove skewing towards low VAF values. 99% simultaneous confidence intervals on frequency are shown, and the total number of calls made is listed below each label. b, Call status of sSNVs called by other methods in LiRA. Calls made by single-cell variant calling methods contain many variants filtered as FPs in LiRA. Accepting only PASS mutations after VQSR in GATK does not change this. In SCcaller, lowering α lowers the proportion of the variants identified in LiRA as FPs, but the proportion remains high. 99% simultaneous confidence intervals are shown, and the size of the LiRA-intersection is listed above each bar. c, Comparison of sSNV types between LiRA FPs and LiRA calls. Well-supported LiRA FPs, distinguished as those that are marked as ‘PASS’ by GATK, differ significantly from LiRA calls in mutational spectra. 99% simultaneous confidence intervals are shown.
References
-
- Hou Y et al. Single-Cell Exome Sequencing and Monoclonal Evolution of a JAK2-Negative Myeloproliferative Neoplasm. Cell 148, 873–885 (2012). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
