

Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis
- PMID: 30888834
- PMCID: PMC6515867
- DOI: 10.1164/rccm.201901-0145OC
Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis
Abstract
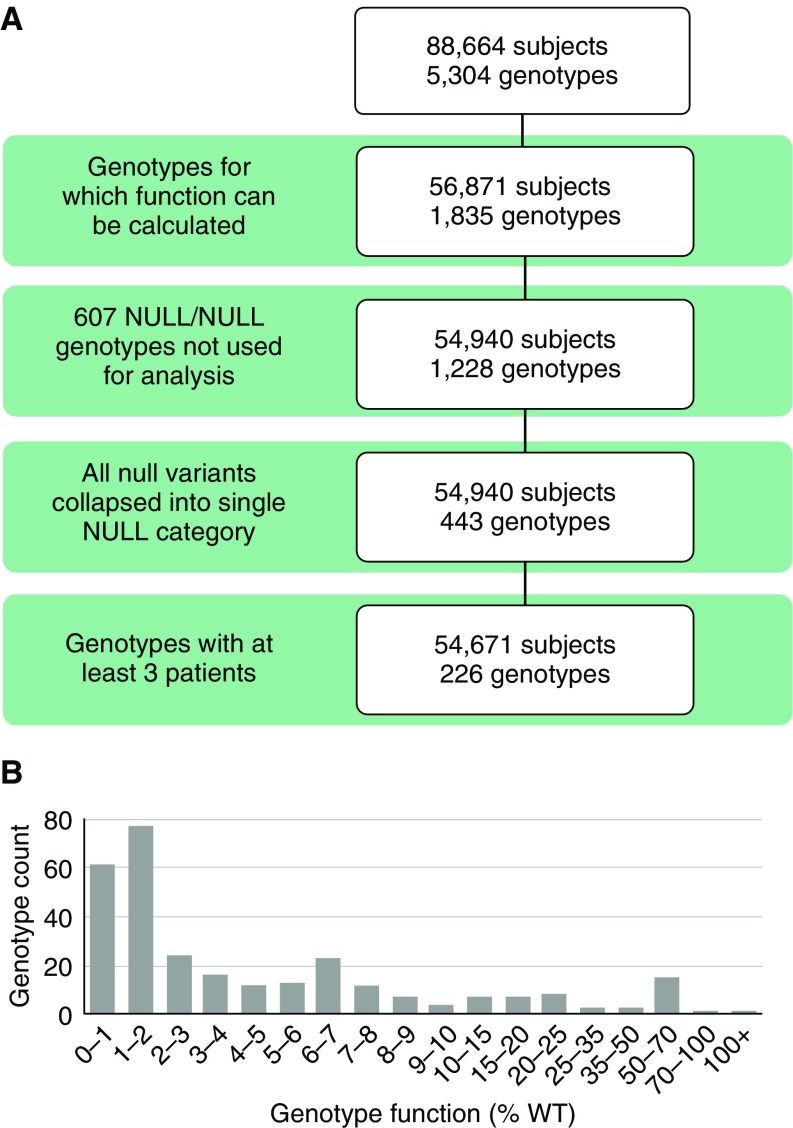
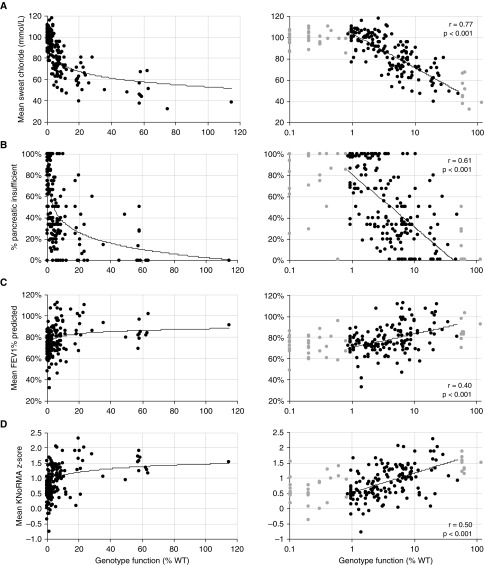
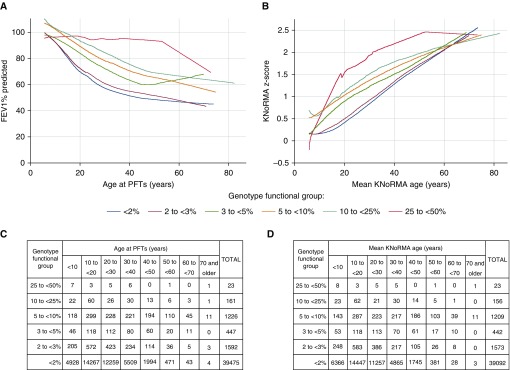
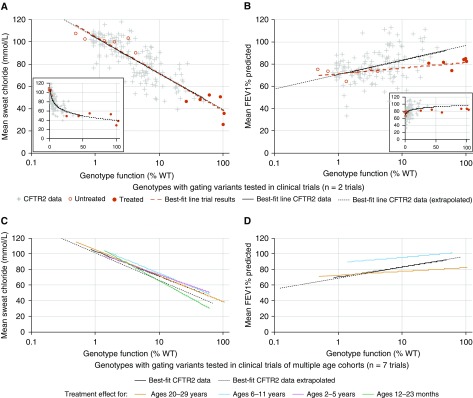
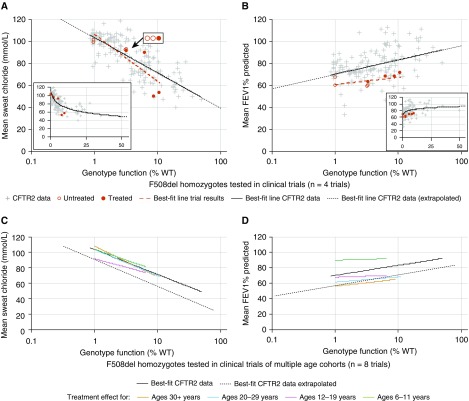
Rationale: The advent of precision treatment for cystic fibrosis using small-molecule therapeutics has created a need to estimate potential clinical improvements attributable to increases in cystic fibrosis transmembrane conductance regulator (CFTR) function. Objectives: To derive CFTR function of a variety of CFTR genotypes and correlate with key clinical features (sweat chloride concentration, pancreatic exocrine status, and lung function) to develop benchmarks for assessing response to CFTR modulators. Methods: CFTR function assigned to 226 unique CFTR genotypes was correlated with the clinical data of 54,671 individuals enrolled in the Clinical and Functional Translation of CFTR (CFTR2) project. Cross-sectional FEV1% predicted measurements were plotted by age at which measurement was obtained. Shifts in sweat chloride concentration and lung function reported in CFTR modulator trials were compared with function-phenotype correlations to assess potential efficacy of therapies. Measurements and Main Results: CFTR genotype function exhibited a logarithmic relationship with each clinical feature. Modest increases in CFTR function related to differing genotypes were associated with clinically relevant improvements in cross-sectional FEV1% predicted over a range of ages (6-82 yr). Therapeutic responses to modulators corresponded closely to predictions from the CFTR2-derived relationship between CFTR genotype function and phenotype. Conclusions: Increasing CFTR function in individuals with severe disease will have a proportionally greater effect on outcomes than similar increases in CFTR function in individuals with mild disease and should reverse a substantial fraction of the disease process. This study provides reference standards for clinical outcomes that may be achieved by increasing CFTR function.
Keywords: cystic fibrosis transmembrane conductance regulator modulator; genotype–phenotype; lung function; sweat chloride.
Figures
Comment in
-
Unraveling the CFTR Function-Phenotype Connection for Precision Treatment in Cystic Fibrosis.Am J Respir Crit Care Med. 2019 May 1;199(9):1053-1054. doi: 10.1164/rccm.201903-0696ED. Am J Respir Crit Care Med. 2019. PMID: 30939246 Free PMC article. No abstract available.
References
-
- Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. - PubMed
-
- Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:1783–1784. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
