

H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis
- PMID: 30890717
- PMCID: PMC6425035
- DOI: 10.1038/s41467-019-09140-x
H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis
Abstract
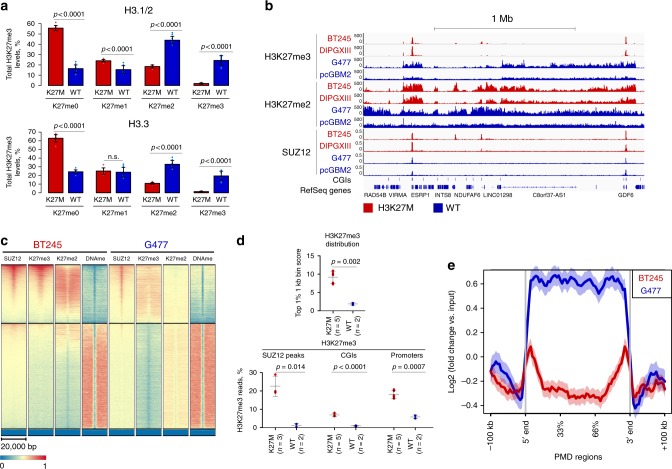
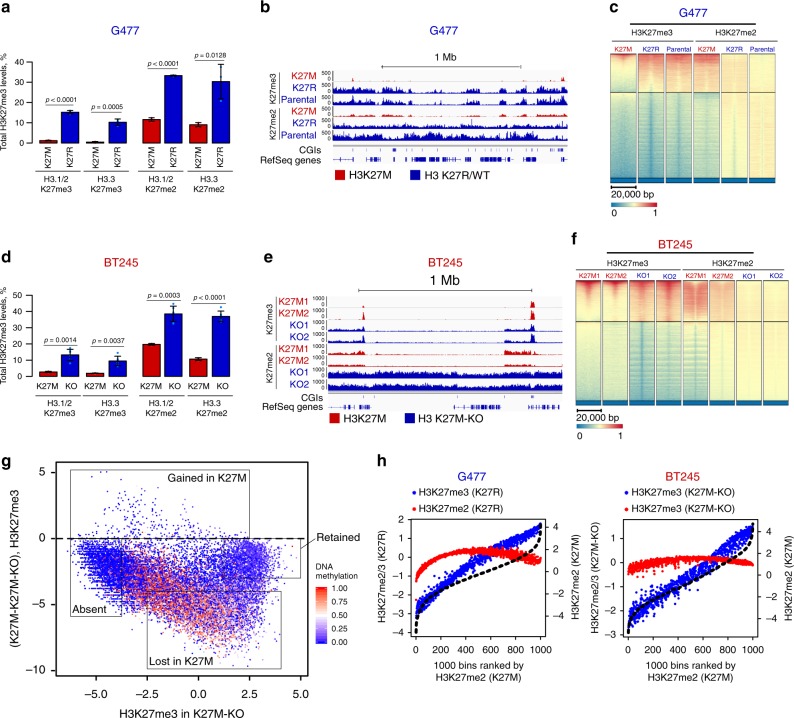
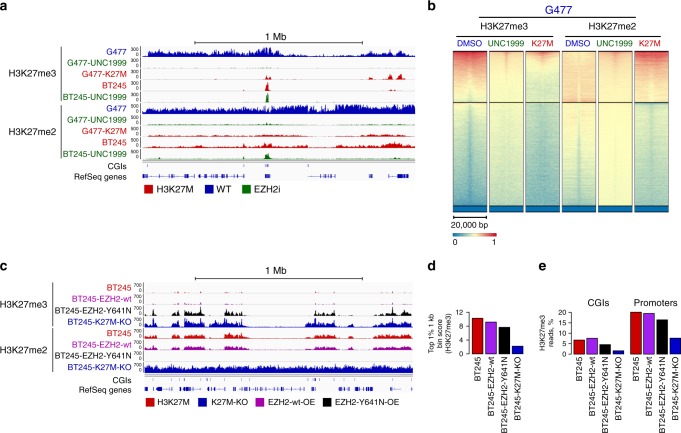
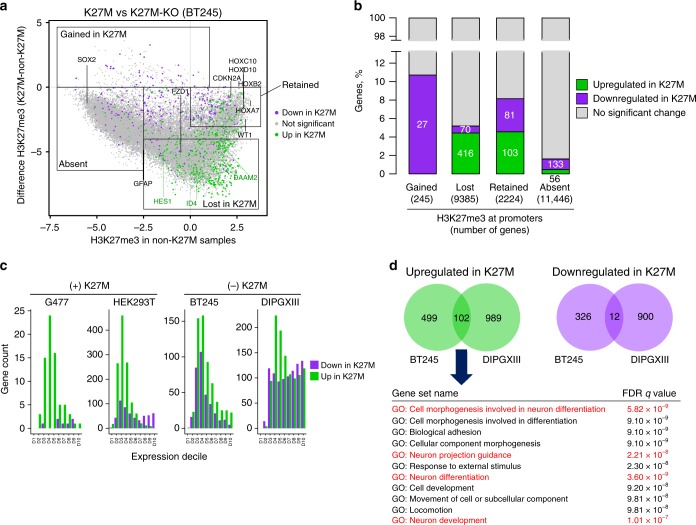
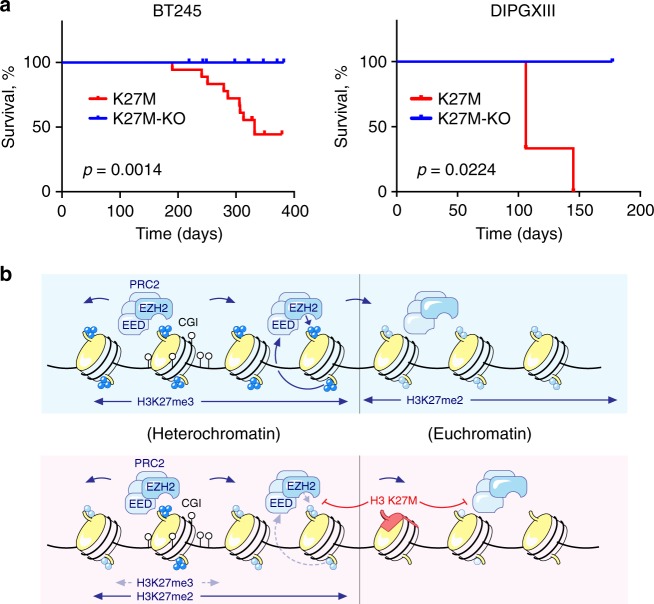
Lys-27-Met mutations in histone 3 genes (H3K27M) characterize a subgroup of deadly gliomas and decrease genome-wide H3K27 trimethylation. Here we use primary H3K27M tumor lines and isogenic CRISPR-edited controls to assess H3K27M effects in vitro and in vivo. We find that whereas H3K27me3 and H3K27me2 are normally deposited by PRC2 across broad regions, their deposition is severely reduced in H3.3K27M cells. H3K27me3 is unable to spread from large unmethylated CpG islands, while H3K27me2 can be deposited outside these PRC2 high-affinity sites but to levels corresponding to H3K27me3 deposition in wild-type cells. Our findings indicate that PRC2 recruitment and propagation on chromatin are seemingly unaffected by K27M, which mostly impairs spread of the repressive marks it catalyzes, especially H3K27me3. Genome-wide loss of H3K27me3 and me2 deposition has limited transcriptomic consequences, preferentially affecting lowly-expressed genes regulating neurogenesis. Removal of H3K27M restores H3K27me2/me3 spread, impairs cell proliferation, and completely abolishes their capacity to form tumors in mice.
Conflict of interest statement
The authors declare no competing interests
Figures
Comment in
-
Demonstrated efficacy and mechanisms of sensitivity of ONC201: H3K27M-mutant diffuse midline glioma in the spotlight.Neuro Oncol. 2024 Jun 3;26(6):991-992. doi: 10.1093/neuonc/noae051. Neuro Oncol. 2024. PMID: 38588460 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
